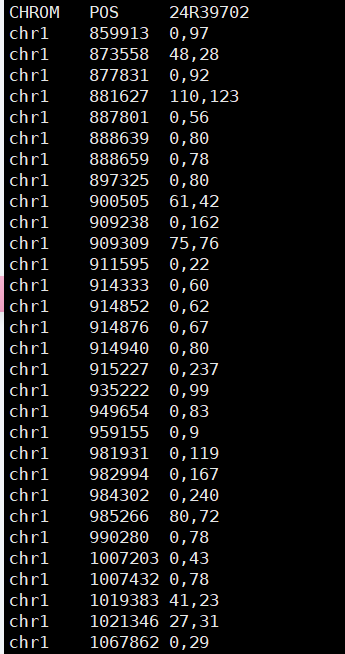
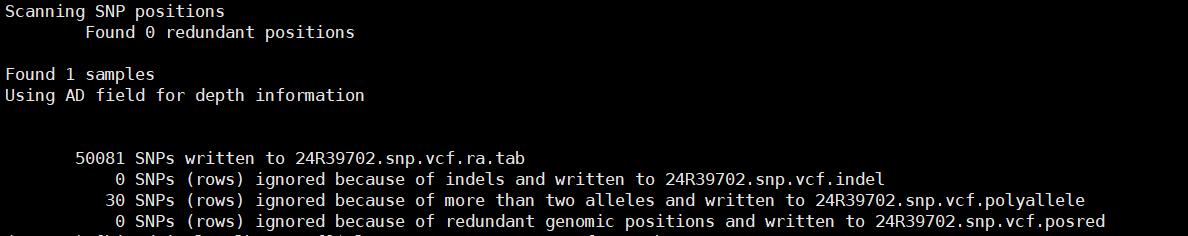
本方法可实现快速提取VCF文件中全部样本的的双等位基因深度数据,并提取各类型数据至Tab分割的文本文件中。 1. 数据处理逻辑 indels去除并单独保存至infile.vcf.indel文件中; SNP非双等位基因的被去除并单独存储至infile.vcf.polyallele文件中; 冗余位点被去除并保存至infile.vcf.posred文件中; ./. 被转换为 0,0 2. 运行示例 python parser_vcf.py sample.vcf *.snp.vcf.ra.tab结果文件: 3. 主脚本程序 # parser_vcf.py #输入








 超级会员免费看
超级会员免费看

 订阅专栏 解锁全文
订阅专栏 解锁全文



















 489
489

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言


 被折叠的 条评论
为什么被折叠?
到【灌水乐园】发言
被折叠的 条评论
为什么被折叠?
到【灌水乐园】发言








