




 详情请看:LiP-MS药物靶点筛选技术
详情请看:LiP-MS药物靶点筛选技术
药物靶点筛选是药物研发和老药新用的核心环节,其目标是识别与疾病相关的关键生物分子靶点,为新药开发和现有药物的再利用提供重要方向。近年来,质谱技术(Mass Spectrometry, MS)在这一领域崭露头角,成为靶点筛选和验证的强大工具。特别是有限蛋白水解质谱技术(Limited Proteolysis Mass Spectrometry, LiP-MS),凭借其高灵敏度、高分辨率的优势,能够直接探测药物与靶点蛋白结合引起的构象变化,从而实现对靶点的高灵敏度和高特异性识别。此外,LiP-MS还可与其他组学技术(如蛋白质组学、代谢组学)相结合,提供更全面的药物作用机制和靶点网络信息,为药物研发提供多维度的数据支持。随着技术的不断优化和应用范围的扩大,LiP-MS有望成为药物研发中不可或缺的关键工具。本文将探讨LiP-MS技术在药物靶点筛选中的应用进展、技术优势及其对未来药物研发的深远影响。
LiP-MS原理
LiP-MS技术,顾名思义,由两部分组成:MS代表质谱(Mass Spectrometry),而LiP即限制性酶解(Limited Proteolysis),这是该技术的核心所在。限制性酶解是指在生理条件下,利用蛋白酶对蛋白质进行有限度的切割。那么,为什么要采用这种方法呢?具体来说,在生理状态下,蛋白质的三维结构使其对酶切具有较强的抗性。为了实现蛋白质的彻底酶切,传统方法通常需要先对蛋白质进行变性处理,使其结构展开,从而暴露出更多的酶切位点。然而,当蛋白质的结构发生改变——例如与小分子药物结合时——这种结合作用会稳定某些酶切位点,减少蛋白质表面肽段的暴露,从而降低它们被蛋白酶切割的可能性。这种结构变化的结果,是生成与原始蛋白质不同的酶切片段。通过分析这些片段的变化,LiP-MS技术能够揭示蛋白质与小分子药物之间的相互作用及其引起的构象变化,为药物靶点筛选提供重要信息。
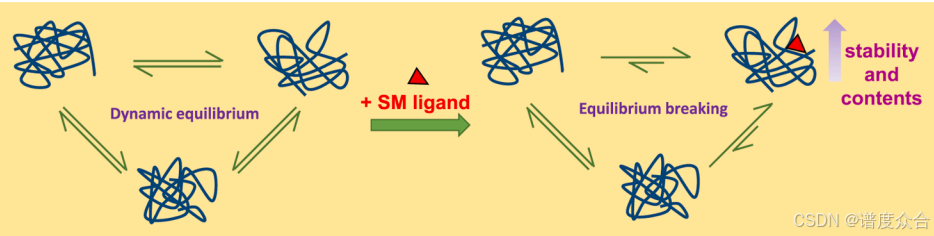
药物稳定蛋白结构
利用这一理论,我们在实验中仅需设置药物处理组与溶剂处理的对照组,并使用非特异性蛋白酶进行处理。这样,药物结合的靶蛋白上将形成酶切位点(肽段)的差异。最终,通过质谱检测,我们可以鉴定并筛选出组间存在差异的肽段(蛋白),从而确认这些为药物作用的结合靶点。这一方法有助于在蛋白质组学平台上实现药物靶点的筛选目标。
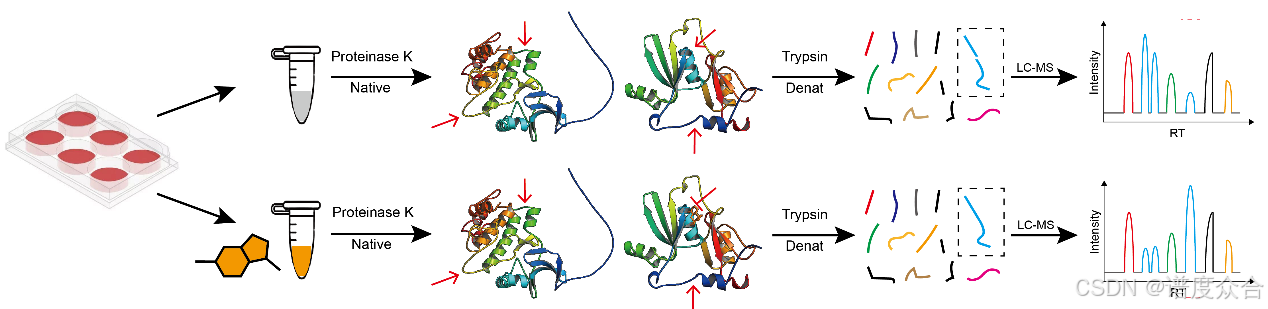
LiP-MS基本流程图
近年来,LiP-MS技术迎来了一项创新性改进,其操作流程如图中所示。该方法在传统LiP-MS流程的基础上进行了优化和扩展,新增了直接使用胰蛋白酶对处理组和对照组样本进行消化的步骤,专门用于蛋白质的定量差异分析。尽管传统LiP-MS流程已能够有效筛选药物靶点,但这一新方法通过整合定量蛋白质组学技术,进一步拓展了其应用范围,使其能够更精准地研究药物在活体环境中的作用机制。当药物作用于细胞时,细胞内的蛋白质表达水平可能发生显著变化,从而影响药物靶点筛选的准确性。为此,新方法通过在药物处理细胞一段时间后再进行靶点筛选,有效校正了肽段量的差异,同时提供了细胞中药物作用机制的详细信息。这一改进不仅提升了LiP-MS技术的可靠性和适用性,还为药物研发提供了更全面的生物学视角。
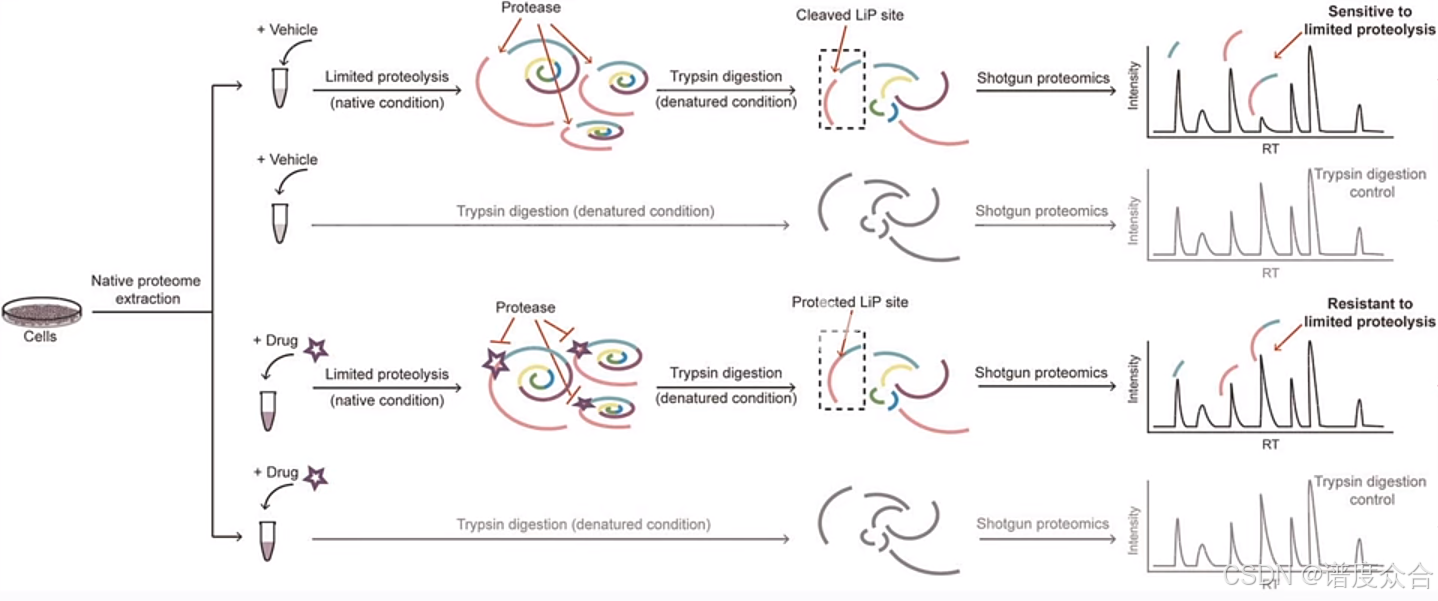
LiP-MS进阶流程图
LiP-MS 技术方面的优势
√全面检测蛋白质信息
LiP-MS技术可以同时检测蛋白质的丰度和结构变化,提供更全面的蛋白质组学信息。
√提供结合序列信息
LiP-MS技术能够提供蛋白质/小分子-蛋白质相互作用的结合序列信息,这对于理解药物作用机制和蛋白质功能状态的变化至关重要。
√无需化学修饰
LiP-MS技术无需对研究的小分子进行化学修饰,这避免了传统化学蛋白质组学方法中可能遇到的分子过大或活性降低的问题。
√适用样本类型广泛
LiP-MS技术适用于多种样本类型,包括蛋白质溶液、组织、细胞等,使其能够在不同的生物系统中发挥重要作用。
√高通量、高分辨率
LiP-MS技术以其高通量和高分辨率的特点,在蛋白质组学研究中具有广泛的应用前景。其对构象变化的识别能力可以达到约12个氨基酸的分辨率,这有助于精确定位小分子的结合位点。
本公司关于LiP-MS代表文章
这是一篇于2024年10月发表在《Theranostics》(影响因子IF为12.4)杂志上的文章,题为“Characterization of dipyridamole as a novel ferroptosis inhibitor and its therapeutic potential in acute respiratory distress syndrome management”。
在该研究中,为了满足客户筛选与双嘧达莫(DIPY)直接结合的靶向蛋白的需求,本公司运用LiP-MS技术协助其识别潜在的药物结合蛋白。通过综合反应组通路分析,我们成功筛选出了关键蛋白——超氧化物歧化酶1(SOD1)。
本公司目前主要着重于药物靶点筛选方面,包含以下三种研究目的场景供大家参考与选择:
| 产品名称 | LiP-MS药物靶点筛选 | LiP-MS药物-蛋白结合域分析 | LiP-MS药物机制研究 |
| 产品目的 | 体外生理条件下寻找小分子药物的潜在作用靶点 | 体外生理条件下研究纯化重组蛋白与目标药物的作用域与结合位点 | 体内给药条件下研究小分子药物的潜在作用靶点及其药物作用机制 |
LiP-MS 送样要求
| 样本类型 | 样本量 | 药物用量要求(非必须) | 运输要求 |
| 细胞 | 1*106 | 摩尔分子质量/10(μg) 或者20μL*5mM的药物溶液 | 干冰运输 |
| 组织 | 5mg | ||
| 蛋白质溶液 | 5μg(浓度不低于0.05μg/μL) |
如需进一步探讨具体的实验样本需求或实验设计,我们欢迎您与我们联系,以便我们能够共同商讨并制定最适合您研究需求的方案。

















 1587
1587

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









