




 更多详情请看:LiP-MS药物靶点筛选技术
更多详情请看:LiP-MS药物靶点筛选技术
现代药物开发面临着高成本、高风险和长周期的挑战,而药物靶点的识别是理解药物作用机制、评估副作用及成药性的关键步骤。小分子药物在疾病治疗中发挥着重要作用,其通过与细胞中特定的靶分子相互作用来发挥功能,因此鉴定小分子药物的靶蛋白对于理解其作用机制和应用至关重要。传统的化学蛋白质组学方法通常需要对小分子进行化学修饰,这可能影响药物的活性构象。相比之下,无标记靶点识别方法(如药物亲和响应靶点稳定性,DARTS)无需化学修饰,逐渐成为研究热点。近年来,蛋白质组学技术,特别是质谱(MS)技术,在药物靶点表征中得到了广泛应用。其中,有限蛋白酶解-质谱法(Limited Proteolysis–Mass Spectrometry, LiP-MS)因其能够高效检测药物结合靶蛋白、解析药物作用机制,在疾病治疗和药物研发中展现出重要价值。这些方法在药物开发中具有广泛的应用前景,尤其是在多靶点或复杂药物系统中,能够提供更全面的靶点蛋白和结合位点信息。接下来,我们将详细介绍DARTS和LiP-MS这两种方法在实验原理、操作步骤及应用场景上的差异,以帮助大家更好地理解这两种技术的独特优势。
1.实验原理不同:
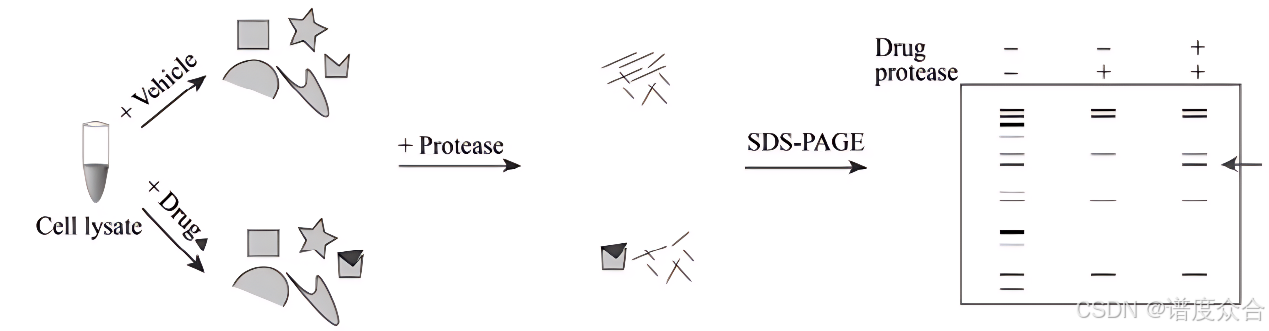
- DARTS:DARTS技术基于小分子药物与靶蛋白结合后,能够降低靶蛋白对蛋白酶的敏感性,从而增强其对蛋白水解酶的抗性。通过SDS-PAGE(十二烷基硫酸钠聚丙烯酰胺凝胶电泳)分离样品并进行染色,可以鉴定出在药物存在下未被水解的蛋白质条带。随后,利用质谱技术对这些受保护的蛋白质条带进行分析,即可准确鉴定出小分子药物的靶标蛋白。

DARTS实验原理图
- LiP-MS:LiP-MS技术通过小分子药物以单一浓度或多个浓度与天然状态下的细胞裂解物共同孵育,随后短暂暴露于非特异性蛋白酶(如蛋白酶K)进行有限酶切。由于小分子药物与蛋白质的结合可能引起蛋白质构象的变化,这种构象变化会影响蛋白酶的切割位点可及性,从而在有限的蛋白酶解过程中产生靶蛋白的特异性片段。接下来,使用胰蛋白酶对这些蛋白质片段进行进一步消化,生成适合定量质谱分析的肽段。通过比较代谢物处理样品与未处理样品中肽段的丰度差异,可以精确识别出与小分子药物结合的蛋白质及其作用位点。
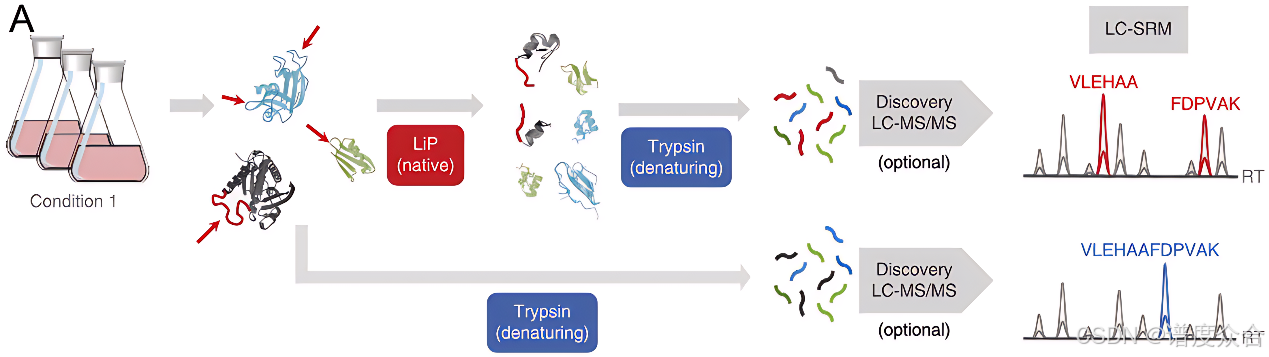
LiP-MS实验原理图
2.实验步骤不同:
- DARTS:
1.细胞裂解;
2.小分子药物与裂解物的孵育;
3.蛋白质酶酶解;
4.通过电泳凝胶染色等方法检测药物处理组和对照组蛋白;
5.鉴定保留的蛋白质条带;
6.对蛋白条带进行质谱,鉴定出药物靶标蛋白。
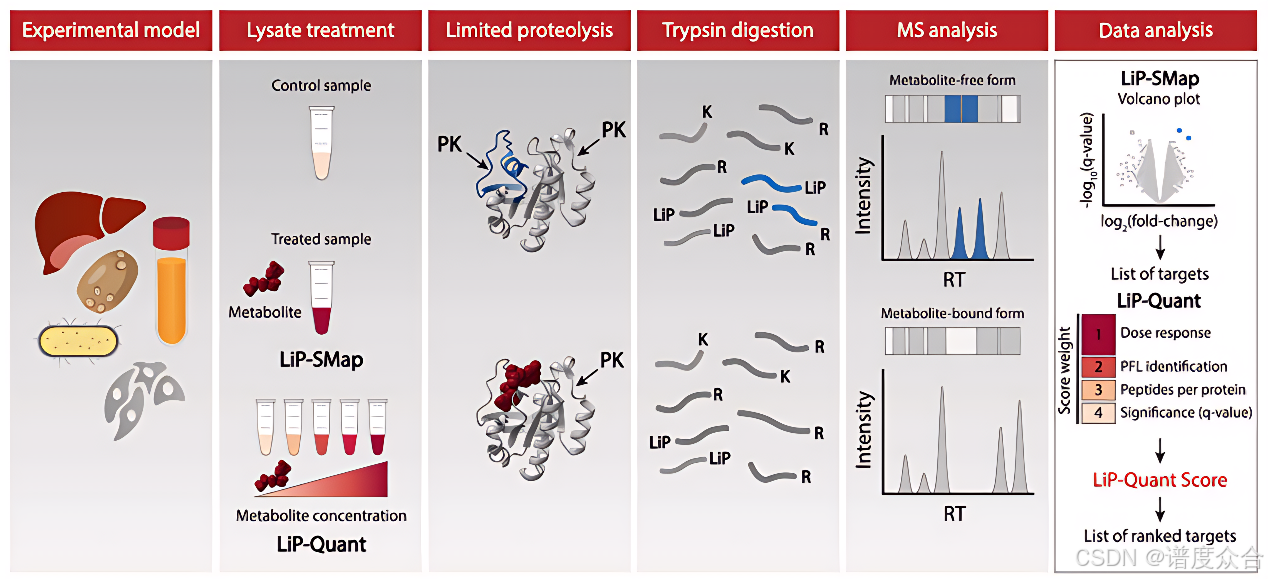
- LiP-MS:
1.细胞裂解;
2.小分子药物与裂解物的孵育;
3.在孵育后的裂解混合物中加入非特异性蛋白酶(如蛋白酶K)进行有限蛋白酶切;
4.使用胰蛋白酶对蛋白质片段进行进一步消化;
5. LC-MS/MS 分析检测肽段的质量和丰度。
3.应用场景不同:
- DARTS:主要应用于小分子药物靶标的鉴定,筛选出与其相互作用的蛋白质;
- LiP-MS:目前已广泛应用于药物靶点发现和脱靶效应筛查、疾病生物标志物的发现、蛋白-蛋白、蛋白-小分子相互作用及网络分析、翻译后修饰与蛋白结构和功能的相关性研究、蛋白质折叠动力学研究、分析蛋白酶活性的变化、新药研发等相关领域。
4.优势及局限性:
- DARTS:
优势:1.DARTS技术无需对蛋白质进行化学修饰,保持了蛋白质的天然状态。
2.DARTS技术能够在单一成分的小分子中鉴定出新的靶标和靶标簇,同时也适用于天然产物和复杂成分的分析。
3.DARTS对蛋白质的纯度要求较低,无需高度纯化的样品。
4.DARTS能够精确识别生物活性小分子与其靶蛋白之间的主要结合基序,为药物作用机制的研究提供了关键信息。
局限性:1.DARTS技术虽然在细胞裂解物中操作简便,但在体内环境中实施仍面临较大挑战。2.传统的DARTS方法依赖于SDS-PAGE和凝胶染色进行目视检测,然而细胞和组织中的大多数蛋白质丰度较低,难以通过无偏倚的蛋白质组学方法进行有效观察。
3.某些蛋白质在天然条件下对蛋白水解酶的敏感性较低,可能影响实验结果的准确性。
- LiP-MS:
优势:1. LiP-MS凭借其高分辨率的特点,能够精确检测小分子与蛋白质的结合位点。
2. LiP-MS能够全面识别和检测多种类型的蛋白质-小分子相互作用,包括变构效应、酶-底物结合、酶-产物复合物以及药物-靶标相互作用。
3. LiP-MS能够在接近生理条件下探测蛋白质的原位结构变化,揭示蛋白质在天然状态下的动态构象特征。
4. LiP-MS适用于多种实验条件,能够检测不同环境(如温度、pH、配体结合等)下蛋白质的结构变化。
5.局限性:
1. 结合位点的确定依赖于质谱(MS)检测到的肽段信息,因此需要靶蛋白具有较高的序列覆盖率,以确保关键区域的充分覆盖和准确鉴定。
2. 蛋白质构象的动态变化可能掩盖或干扰结合位点的识别,增加了数据解析的复杂性,可能影响结合位点的准确定位。
6.总结:DARTS和LiP-MS技术在蛋白质组学研究中具有高度的互补性,能够为蛋白质-小分子相互作用及蛋白质结构研究提供更全面的信息。例如,研究者可以首先利用DARTS技术高效筛选出与小分子结合的潜在靶蛋白,随后通过LiP-MS技术深入分析小分子结合后靶蛋白的构象变化,从而系统揭示小分子的作用机制。这两种技术在药物靶标发现、药物作用机制解析以及蛋白质结构与功能研究等领域展现出巨大的应用潜力。随着技术的不断优化和完善,DARTS和LiP-MS必将在蛋白质组学研究中发挥越来越重要的作用,为生命科学研究提供更加精准和强大的工具。
作为新兴的蛋白质组学技术,DARTS和LiP-MS在蛋白质-小分子相互作用研究和蛋白质结构动态解析方面具有独特的优势。它们的出现不仅拓展了蛋白质组学的研究维度,也为相关领域提供了全新的研究思路和方法。未来,随着技术的进一步发展,DARTS和LiP-MS有望在基础研究和药物开发中发挥更大的价值,推动蛋白质组学研究迈向新的高度。
详情请看:LiP-MS药物靶点筛选技术

















 2463
2463

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









