







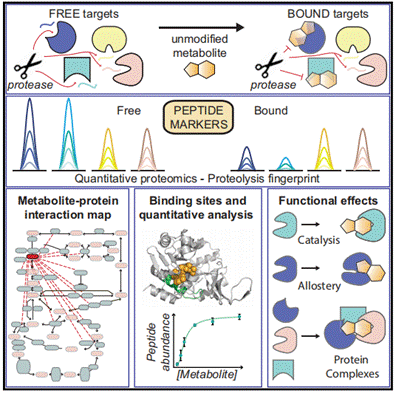
有限蛋白水解质谱技术(Limited Proteolysis Mass Spectrometry,LiP-MS)是一种结合蛋白质有限酶解与质谱分析的技术,通常用于研究小分子化合物(代谢物、天然产物等)与蛋白质之间的相互作用。此外,还有研究利用该技术解析蛋白质结构动态变化、鉴定蛋白质-配体结合位点及相互作用区域。
Piazza I, et al. Cell. 2018.
LiP-MS技术原理
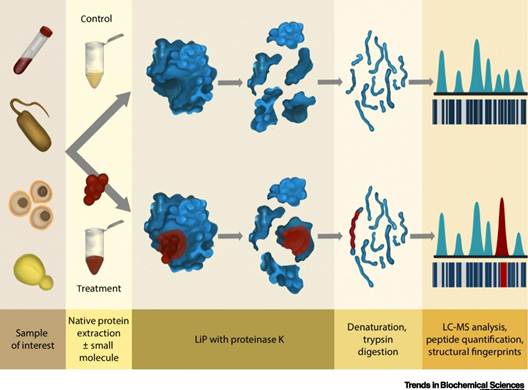
LiP-MS技术由苏黎世联邦理工学院研究团队发明,该技术的核心原理在于药物与靶蛋白结合后会改变靶蛋白的空间构象/稳定性/产生位阻变化,从而会掩盖一些蛋白质表面的酶切位点。所以利用蛋白酶对蛋白质表面暴露区域的选择性酶切,结合质谱对酶切肽段的分析,就能够揭示蛋白质构象变化或结合事件中的结构信息。
Piazza I, et al. Cell. 2018.
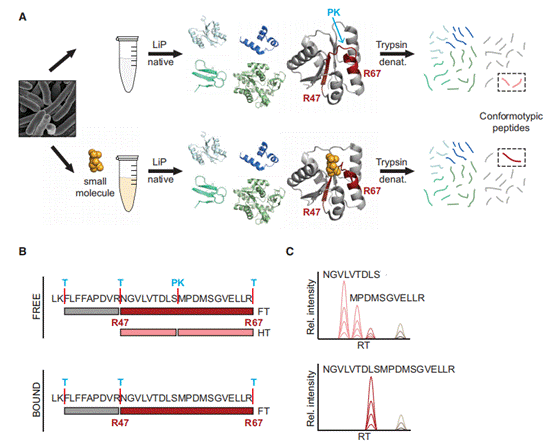
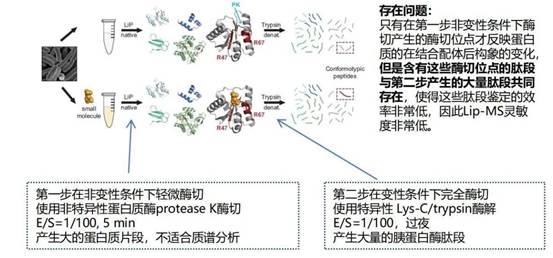
LiP-MS技术的原理在于药物与靶蛋白结合后会改变靶蛋白的空间构象/稳定性/产生位阻变化,从而会掩盖一些蛋白质表面的酶切位点。基于此原理鉴定靶蛋白需要进行两步酶切:第一步使用非特异性的酶(通常采用PK酶)进行酶切,并限制酶对蛋白水解的持续时间,PK酶主要靶向和标记表面暴露和柔性的蛋白质区域,由于配体结合会导致蛋白表面可及性和局部结构灵活性改变,而这会影响蛋白的消化效率;第二步则使用胰酶进行进一步酶切,将第一步的酶切产物酶解成适于质谱分析的小肽段。
Lip-MS应用存在的问题与误区
然而这种鉴定方式的问题在于:只有在第一步酶切产生的酶切位点才能够反映蛋白质在结合药物后构象的变化,但是含有这些酶切位点的肽段与第二步胰酶酶解产生的大量肽段共同存在。
所以,在LiP-MS的体系中包含着完全胰蛋白酶消化肽段(蛋白质按照胰蛋白酶的切割位点规则被充分地切割成特定长度和序列的肽段)和半胰蛋白酶消化肽段(由于蛋白质的折叠状态、配体结合等因素影响,导致蛋白质被保护或暴露其特定切割位点,影响了蛋白的水解而产生的消化不完全的肽段)。想要在这种复杂的体系中从肽段回归到蛋白,增加了数据分析的难度、复杂性以及分析维度,导致对靶点蛋白的鉴定效率低,灵敏度有限。
LiP-MS存在的这一问题在一些靶点相关研究中也有所提及和验证。LiP-MS此前只做大肠杆菌等简单样品,直到2020年在真核细胞样品中测试了LiP-MS鉴定药物靶标的性能,但是测试结果发现,在一个药物剂量下LiP-MS不能区分真的鉴定与假的鉴定,这一结果说明,LiP-MS在靶蛋白的鉴定方面性能较差。除此之外,在这种复杂的体系且有限时间内,MS鉴定的效率也是有限的,并且会优先检测高丰度蛋白,并会掩盖低丰度信号(第一步酶切产生的多肽),导致低丰度蛋白检出结果实际较差,也就是LiP-MS结果中大多数都是高丰度蛋白,而忽略的低丰度蛋白中很有可能正是药物结合发挥关键作用的调控型蛋白。

升级版LiP-MS——LiP-Quant技术
也有研究为了解决这一问题,在LiP-MS基础上,采用了基于机器学习的框架,并利用药物多浓度剂量来识别药物靶标(即LiP-Quant技术)。此外,作者基于机器学习建立了一套综合得分体系,旨在筛选出药物真正结合的靶点蛋白。经测验,LiP-Quant能够优先确定真正的药物靶标并且减少假阳性鉴定,相比于LiP-MS,其灵敏度与特异性大大提高。

Piazza I, et al. Nat Commun. 2020.
然而在LiP-Quant实验中需要细胞裂解液与至少7个剂量的药物共孵育,随后采用PK酶进行限制性酶切,靶蛋白中至少结合位点的酶切特征会由于给药而改变,真正的靶肽段会随着药物剂量丰度随之变化。相比于LiP-MS其实验复杂度大大增加,实验时间和成本也随之提高。此外,LiP-Quant筛选靶蛋白的评分体系使其数据分析的难度变得更高,定量计算更为复杂。尽管其鉴定性能要明显高于LiP-MS,但对低丰度蛋白的检测仍然受限。
LiP-MS鉴定结合位点存在的局限
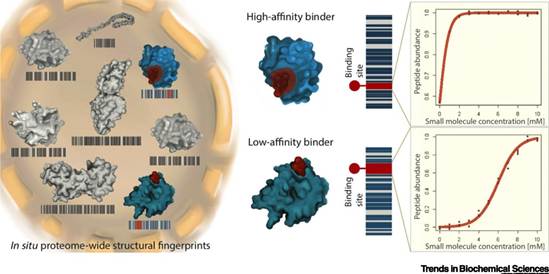
还有一些研究表明,LiP-MS技术能够根据差异肽段映射至蛋白的三维结构,由此鉴定配体与蛋白的结合位点。但是,在这一方面仍存在一些不可忽视的局限性:
- 但是通过质谱来检测结合界面处的肽段,这需要靶蛋白具有较高的序列覆盖率,才能确保关键区域的充分覆盖和准确鉴定。若结合位点所在的肽段区域未被覆盖,质谱无法捕捉到这些区域的结构变化(如药物结合导致的构象变化),则会遗漏或无法准确识别该结合位点。但目前的质谱技术中肽段覆盖率尤其是大蛋白的肽段覆盖率其实是比较低的,并不能做到全蛋白氨基酸逐一测序,所以该方法仍然更偏向于中/高丰度蛋白。
- 在LiP-MS的体系中包含完全胰蛋白酶消化肽段和半胰蛋白酶消化肽段,通过这两种肽段的上/下调来判断结合区域,但是无法准确的区分完全胰蛋白酶消化肽段和半胰蛋白酶消化肽段,所以仍会造成结合区域不清晰的问题。
- 此外,构象变化也会阻碍结合位点的识别,如果配体在结合区域之外引起蛋白结构变化,则可以检测到相互作用,但无法准确确定结合位点。
Pepelnjak M, et al. Trends Biochem Sci. 2020.
LiP-MS鉴定结果
由于LiP-MS技术要在复杂体系中由肽段回归到靶点蛋白,这需要精细的分析比较差异多肽的酶切位点,但是这种分析难度大,复杂性和分析维度高,所以目前多数文章中的分析方式仍是通过特征性肽/代表性肽来判断配体结合的靶点蛋白。
在一些研究中发现,在LiP-MS实验中,药物与靶蛋白的结合可能通过物理遮挡或构象稳定化影响蛋白酶对靶蛋白的切割效率,导致部分肽段无法生成,从而干扰靶点的鉴定。若药物分子量极大(如抗体药物)或结合位点完全覆盖蛋白酶切位点,这种较为极端的情况可能导致靶蛋白所有潜在切割位点均被保护,且无其他构象变化区域,可能完全阻断后续酶切,导致相应肽段缺失。
在结合鉴定方面,大多数文献的结果中仅对Lip-MS提供的差异蛋白层进行统计,几乎没有文章通过Lip-MS的结果去分析小分子与多肽结合位点。并且这些蛋白的上下调是由某个蛋白多条多肽的定量数据平均化计算获得,对于蛋白靶点是否被小分子保护的确认并不准确,尤其在一个蛋白同时存在上下调的多肽时,蛋白靶点的确认过程中,假阴性非常高。
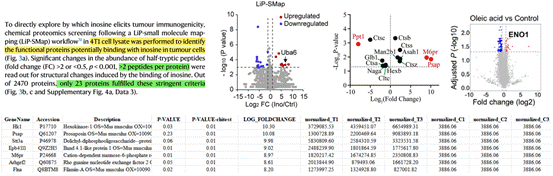
总的来说,LiP-MS技术筛选药物靶点存在着以下几点问题:
- PK酶切割,使肽段反映蛋白构象变化,再利用胰酶将蛋白酶解成适合MS分析的肽段,两步酶解产生的肽段共同存在,导致低丰度蛋白检出结果实际较差;
- 鉴定结合区域涉及到蛋白序列覆盖率、完全胰蛋白酶消化肽段和半胰蛋白酶消化肽段共同存在等问题,导致遗漏或无法准确识别结合位点;
- 鉴定效率低、灵敏度低;
- 数据分析复杂、难度大、分析维度高。
LiP-MS技术能够提供对小分子靶点筛选和蛋白质构象变化的新见解,但是其主要的适用场景还是大肠杆菌等蛋白质组复杂度低的体系,因为此类简单体系的背景干扰少,组分简单,后续MS解析和数据处理的难度更低,准确性更高。此外,LiP-MS技术还更适用于代谢物或分子量较小的小分子药物筛选靶点,因为这类样本结构较为简单,与靶蛋白的结合模式更为直接、简单,利于后续质谱鉴定其结合靶点。
随着技术的发展进步,还有许多研究者在原有的LiP-MS技术原理和步骤基础上不断地调整和优化,现已发展到了解疾病机制,识别疾病标志物的应用中来。虽然已经取得了一些成功,但是其面临的挑战仍然存在,未来的研究更可能聚焦于天然蛋白质研究、更精确的肽鉴定和结构解释、以及识别特异性蛋白质变化进行高通量结构蛋白分析等。其与前沿技术的融合、智能化分析能力的提升,以及在复杂场景中的应用拓展,将有机会为精准医疗和创新药物开发带来新的进展。

















 1936
1936

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









