







三、反应动力学
L12 稀溶液中的反应动力学
目录
1. 背景介绍
在研究电化学能量系统时,非平衡态的首要关注点是反应动力学。本次讲座中,我们将从通用的过渡态理论开始,并将其应用到电极上的法拉第电荷转移反应。
2. 反应速率的随机理论
2.1 过渡态理论
反应动力学的经典过渡态理论基于由分子间力引起的随机过程。反应复合物通过热激发扩散穿越一个能量景观 U ( x ) U(x) U(x)(关于反应坐标的函数)以偶尔越过“过渡态”处的能垒 U T S U_{TS} UTS,该能垒分隔了初态和末态 U 1 U_1 U1和 U 2 U_2 U2。该能垒能量需要大于热电压,以保证转变是“稀有事件”而不是常见的热波动:
Δ U 1 , 2 = U T S − U 1 , 2 ≫ k B T \Delta U_{1,2} = U_{TS} - U_{1,2} \gg k_B T ΔU1,2=UTS−U1,2≫kBT
这样可以确保反应物和产物在能量极小值附近维持在定义明确、寿命较长的“状态”中。
在稀溶液极限下,能量屏障的“逃逸”问题可以进行详细分析,此时反应物遵循独立的随机过程(随机游走),不会改变能量景观。每个分子的平均反应速率 r r r(事件/时间),等于从极小值到过渡态的平均首次通过时间 τ \tau τ的倒数:
r = 1 τ ∼ ν e − Δ U / k B T r = \frac{1}{\tau} \sim \nu e^{-\Delta U/k_B T} r=τ1∼νe−ΔU/kBT
在渐近极限 Δ U ≫ k B T \Delta U \gg k_B T ΔU≫kBT下,这是任何热激活过程的熟悉的Arrhenius温度依赖性。该结论也可以通过这样推导出:反应速率与系统在过渡态处的Boltzmann分布的概率成正比, p ∝ e − Δ U / k B T p \propto e^{-\Delta U/k_B T} p∝e−ΔU/kBT。
前因子 ν \nu ν取决于能量景观在极小值处的曲率 K m i n = U m i n ′ ′ > 0 K_{min} = U''_{min} > 0 Kmin=Umin′′>0和在过渡态(鞍点)处的曲率 K T S = − U T S ′ ′ > 0 K_{TS} = -U''_{TS} > 0 KTS=−UTS′′>0,例如对于从对称井中的逃逸,前因子( ν − ( K m i n , K T S ) \nu-(K_{min},K_{TS}) ν−(Kmin,KTS))可以表示为:
ν = 2 D 0 K m i n K T S π k B T ( 1 + a k B T Δ U + b ( k B T ) 2 Δ U 2 + ⋯ ) \nu = \frac{\sqrt{2 D_0 K_{min} K_{TS}}}{\pi k_B T} \left( 1 + \frac{a k_B T}{\Delta U} + \frac{b (k_B T)^2}{\Delta U^2} + \cdots \right) ν=πkBT2D0KminKTS(1+ΔUakBT+ΔU2b(kBT)2+⋯)
其中 D 0 D_0 D0是表征分子尺度热噪声的扩散系数,常数 a a a和 b b b依赖于能量景观的更高阶导数。
通过将极小值和过渡态鞍点区域的宽度分别定义为 L m i n L_{min} Lmin和 L T S L_{TS} LTS,我们可以将前因子( ν − ( L m i n , L T S ) \nu-(L_{min},L_{TS}) ν−(Lmin,LTS))表示为:
ν = D 0 L m i n L T S ( Δ U k B T ) 3 / 2 ( 1 + a k B T Δ U + b ( k B T ) 2 Δ U 2 + ⋯ ) \nu = \frac{D_0}{\sqrt{L_{min} L_{TS}}} \left( \frac{\Delta U}{k_B T} \right)^{3/2} \left( 1 + \frac{a k_B T}{\Delta U} + \frac{b (k_B T)^2}{\Delta U^2} + \cdots \right) ν=LminLTSD0(kBTΔU)3/2(1+ΔUakBT+ΔU2b(kBT)2+⋯)
对于狭窄的势阱( L m i n L_{min} Lmin小),反应速率增强,因为弹簧常数较大,振动频率更高,导致更多的逃逸尝试。同样,在过渡态周围的鞍点较窄时,逃逸过程会浪费更少的尝试在鞍点区域徘徊并返回原来的井中。
3. 稀溶液中的反应
考虑反应 ∑ s i R i → ∑ s j P j \sum s_i R_i \rightarrow \sum s_j P_j ∑siRi→∑sjPj,其中反应物处于状态1,产物处于状态2。对于每个从状态1到状态2的反应复合物,都存在一个能量为 U T S U_{TS} UTS的过渡态。状态能量为:
U 1 = ∑ s i , 1 U i , 1 U_1 = \sum s_{i,1} U_{i,1} U1=∑si,1Ui,1
U 2 = ∑ s j , 2 U j , 2 U_2 = \sum s_{j,2} U_{j,2} U2=∑sj,2Uj,2
对于正向(或反向)反应,每单位体积的这种跃迁次数与反应物浓度成正比。可以计算净反应速率(每个反应位点的反应次数/时间):
R
=
R
1
→
2
−
R
2
→
1
=
ν
1
c
1
e
−
(
U
T
S
−
U
1
)
/
k
B
T
−
ν
2
c
2
e
−
(
U
T
S
−
U
2
)
/
k
B
T
R = R_{1 \rightarrow 2} - R_{2 \rightarrow 1} = \nu_1 c_1 e^{-(U_{TS} - U_1) / k_B T} - \nu_2 c_2 e^{-(U_{TS} - U_2) / k_B T}
R=R1→2−R2→1=ν1c1e−(UTS−U1)/kBT−ν2c2e−(UTS−U2)/kBT
【Note:1→2:
Δ
G
1
→
T
S
=
U
T
S
−
U
1
\Delta G_{1\to\mathrm{TS}}=U_{\mathrm{TS}}-U_1
ΔG1→TS=UTS−U1】
正负反应速率的比值为:
R 1 → 2 R 2 → 1 = ν 1 c 1 ν 2 c 2 e ( U 1 − U 2 ) / k B T \frac{R_{1 \rightarrow 2}}{R_{2 \rightarrow 1}} = \frac{\nu_1 c_1}{\nu_2 c_2} e^{(U_1 - U_2) / k_B T} R2→1R1→2=ν2c2ν1c1e(U1−U2)/kBT
在平衡状态下,这一比值为1,两边同时取对数得:
Δ U e q . = ( U 1 − U 2 ) e q . = Δ U 0 + k B T ln c 2 c 1 \Delta U^{\mathrm{eq.}}=(U_1-U_2)^{\mathrm{eq.}}=\Delta U^0+k_BT\ln\frac{c_2}{c_1} ΔUeq.=(U1−U2)eq.=ΔU0+kBTlnc1c2
其中 Δ U 0 = k B T ln ( ν 2 ν 1 ) \Delta U_0 = k_B T \ln \left( \frac{\nu_2}{\nu_1} \right) ΔU0=kBTln(ν1ν2),这看起来类似于能斯特方程。
4. 稀溶液中的法拉第反应
4.1 初态和末态的标准形式
考虑一般的半电池法拉第反应,我们可以以标准形式写为:
∑ s i M i z i → n e − ∑ j s R j R j z R , j → ∑ i s O i O i Z 0 , i + n e − \sum s_i M_i^{z_i} \rightarrow ne^-\\ \sum_js_{R_j}R_j^{z_{R,j}}\to\sum_is_{O_i}O_i^{Z_{0,i}}+ne^- ∑siMizi→ne−j∑sRjRjzR,j→i∑sOiOiZ0,i+ne−
该反应在正向方向上生成 n n n个电子(氧化反应)。我们将其分为还原态的反应物( s i > 0 s_i > 0 si>0)和氧化态的产物( s i < 0 s_i < 0 si<0)。通过电荷守恒,我们有:
∑ s O i Z O i − n = ∑ s R j z R j \sum s_{Oi} Z_{Oi} - n = \sum s_{Rj} z_{Rj} ∑sOiZOi−n=∑sRjzRj
初态和末态的能量可以表示为:
U 1 = U R = ∑ s R , j [ U R , j 0 + z R , j e Φ ] = U R 0 + q R Φ U_1 = U_R = \sum s_{R,j} \left[ U^0_{R,j} + z_{R,j} e \Phi \right] = U^0_R + q_R \Phi U1=UR=∑sR,j[UR,j0+zR,jeΦ]=UR0+qRΦ
U
2
=
U
O
=
∑
s
O
,
i
[
U
O
,
i
0
+
z
O
,
i
e
Φ
]
=
U
O
0
+
q
O
Φ
U_2 = U_O = \sum s_{O,i} \left[ U^0_{O,i} + z_{O,i} e \Phi \right] = U^0_O + q_O \Phi
U2=UO=∑sO,i[UO,i0+zO,ieΦ]=UO0+qOΦ
其中,
q
o
=
∑
s
O
i
Z
O
i
q_{o}=\sum s_{O_{i}}Z_{O_{i}}
qo=∑sOiZOi且
q
R
=
∑
s
R
j
z
R
j
q_{R}=\sum s_{R_{j}}z_{R_{j}}
qR=∑sRjzRj.
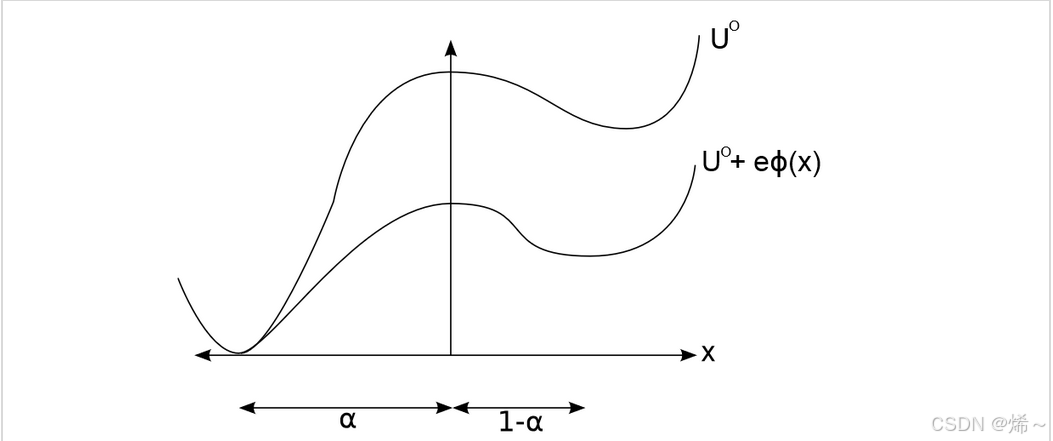
4.2 过渡态的Butler-Volmer模型
Butler-Volmer假设过渡态的静电能是氧化态和还原态静电能的加权平均:
U T S = U T S 0 + α q R Φ + ( 1 − α ) ( q O Φ − n e Φ e ) U_{TS} = U^0_{TS} + \alpha q_R \Phi + (1 - \alpha) \left( q_O \Phi - ne \Phi_e \right) UTS=UTS0+αqRΦ+(1−α)(qOΦ−neΦe)
其中 α \alpha α是传递系数(对称因子),表示过渡态处还原态静电能的权重。通常假设对称电子转移,即 α = 0.5 \alpha = 0.5 α=0.5。
对于电极电位,我们可以得到净反应速率为:
R = k a ∏ i c i , R s i , R e − [ α q R Φ + ( 1 − α ) ( q O Φ − n c Φ e ) − q R Φ ] / ( k T ) − k c ∏ i c i , O s i , O e − [ α q R Φ + ( 1 − α ) ( q O Φ − n c Φ e ) − q O Φ ] / ( k T ) R=k_a\prod_ic_{i,R}^{s_{i,R}}e^{-[\alpha q_R\Phi+(1-\alpha)(q_O\Phi-nc\Phi_e)-q_R\Phi]/(kT)}-k_c\prod_ic_{i,O}^{s_{i,O}}e^{-[\alpha q_R\Phi+(1-\alpha)(q_O\Phi-nc\Phi_e)-q_O\Phi]/(kT)} R=kai∏ci,Rsi,Re−[αqRΦ+(1−α)(qOΦ−ncΦe)−qRΦ]/(kT)−kci∏ci,Osi,Oe−[αqRΦ+(1−α)(qOΦ−ncΦe)−qOΦ]/(kT)
其中我们将
U
i
O
U_i^O
UiO 吸收为阳极(氧化)和阴极(还原)反应速率常数,分别为
k
a
k_a
ka 和
k
c
k_c
kc。
利用电荷守恒
q
O
−
q
R
=
n
q_O − q_R = n
qO−qR=n,我们最终将稀溶液中的法拉第反应速率的一般形式
其中:
R
=
k
a
∏
i
c
i
,
R
s
i
,
R
e
(
1
−
α
)
n
e
Δ
Φ
/
(
k
B
T
)
−
k
c
∏
i
c
i
,
O
s
i
,
O
e
−
α
n
e
Δ
Φ
/
(
k
B
T
)
R=k_a\prod_ic_{i,R}^{s_{i,R}}e^{(1-\alpha)ne\Delta\Phi/(k_BT)}-k_c\prod_ic_{i,O}^{s_{i,O}}e^{-\alpha ne\Delta\Phi/(k_BT)}
R=kai∏ci,Rsi,Re(1−α)neΔΦ/(kBT)−kci∏ci,Osi,Oe−αneΔΦ/(kBT)
Δ
Φ
=
Φ
e
−
Φ
\Delta \Phi = \Phi_e - \Phi
ΔΦ=Φe−Φ
表示电极电位与溶液电位的差值。
补:
-
阿伦尼乌斯方程
-
阿伦尼乌斯方程描述了化学反应速率常数 ( k ) 与温度之间的关系:
k = A e − E a / R T k = A e^{-E_a / RT} k=Ae−Ea/RT
-
k k k:反应速率常数,表示化学反应的速率。
-
A A A:频率因子,也称为前指数因子,代表分子有效碰撞的频率。
-
E a E_a Ea:活化能,单位通常为 J/mol 或 kJ/mol,表示反应物分子必须克服的能量障碍。
-
R R R:气体常数,( R = 8.314 , \text{J/(mol·K)} )。
-
T T T:温度,以开尔文(K)为单位。
-
线性化形式
-
通过对阿伦尼乌斯方程两边取自然对数,可以得到线性化形式:
ln k = ln A − E a R T \ln k = \ln A - \frac{E_a}{R T} lnk=lnA−RTEa
- 如果绘制 ln k \ln k lnk对 1 T \frac{1}{T} T1 的图(阿伦尼乌斯图),斜率为 − E a R -\frac{E_a}{R} −REa,从中可以确定活化能 E a E_a Ea。
5. 总结
- 过渡态理论用于描述反应速率,基于热激发穿越能垒的随机过程。
- 稀溶液中的反应可以通过Arrhenius关系描述,反应速率与过渡态能量相关。
- 法拉第反应的Butler-Volmer模型描述了过渡态的静电能为还原态和氧化态的加权平均。
- 反应速率的计算涉及电荷守恒和反应物及产物的浓度关系,类似于能斯特方程。

















 2109
2109

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









