







三、反应动力学
L14 浓溶液中的法拉第反应
目录
1. 浓溶液中的反应
稀溶液中的化学动力学-假设:正反应和逆反应的速率与浓度成正比
浓溶液中的化学动力学-假设:引入超额化学势,其描述了与稀溶液中不同的额外相互作用,例如排斥体积效应和外部系统的贡献。
1.1 超额化学势
超额化学势 μ i e x \mu_i^{ex} μiex定义为:
μ i = k B T ln C i + μ i e x \mu_i = k_B T \ln C_i + \mu_i^{ex} μi=kBTlnCi+μiex
其中 C i C_i Ci是状态 i i i的浓度。超额化学势 μ i e x \mu_i^{ex} μiex与活度系数 γ i \gamma_i γi有关:
μ i e x = k B T ln γ i \mu_i^{ex} = k_B T \ln \gamma_i μiex=kBTlnγi
活度系数 γ i \gamma_i γi描述了与理想溶液行为的偏离。对于具体的例子,例如晶格气模型,化学势是基于每个晶格位置定义的,其中 k B T ln C i k_B T \ln C_i kBTlnCi表示稀薄、非相互作用气体的熵化学势,所有的相互作用自由能都包含在 μ i e x \mu_i^{ex} μiex中。
在这种解释中, μ i \mu_i μi代表扩散化学势(diffusional chemical potential),即在一定条件下(例如固定格点数)为使系统达到状态 i i i所需的总自由能变化。这不同于真正的热力学化学势,后者仅涉及增加粒子时的自由能变化而不改变系统其他条件。
例如,对于晶格气系统中每个晶格位置的化学势,我们可以表示为:
μ i = k B T ln c − k B T ln ( 1 − c ) = μ particle − μ vacancy \mu_i = k_B T \ln c - k_B T \ln (1 - c) = \mu_{\text{particle}} - \mu_{\text{vacancy}} μi=kBTlnc−kBTln(1−c)=μparticle−μvacancy
其中,第一项描述了生成粒子的自由能变化,第二项描述了生成空位的自由能变化。
1.2 净反应速率的推导
考虑从两个准平衡态(自由能井)之间通过过渡态的跃迁。图1描述了一个概念性的反应图示,展示了离开平衡态的过程中超额化学势的景观。
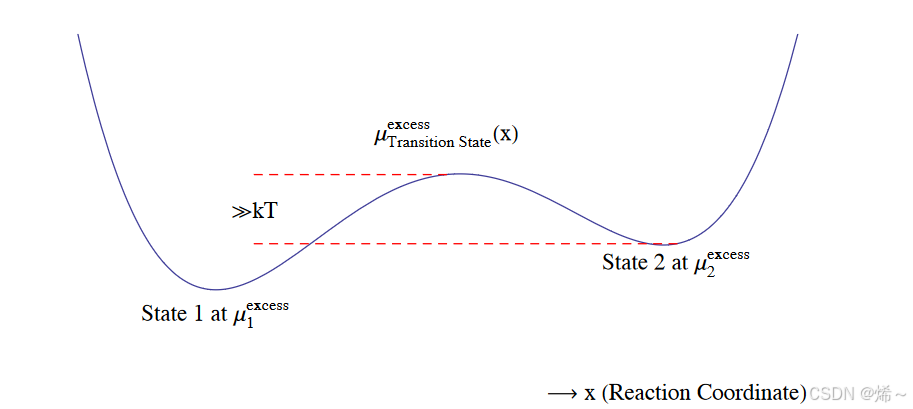
图 1:非平衡过程的概念反应图。示意性能量函数旨在表示过量化学势的景观,同时考虑到成分效应和对能量景观的其他贡献。
设定反应复合物在超额化学势景观中随机行走,我们使用第一通过时间统计理论来描述反应速率。设定从状态1到过渡态的平均首次通过时间为 τ 1 → T S \tau_{1\to TS} τ1→TS,则单位时间内找到反应物在状态1并向前跃迁的概率为:
1 τ 1 → T S ∝ e − ( μ T S e x − μ 1 e x ) / k B T \frac{1}{\tau_{1\to TS}} \propto e^{- (\mu_{TS}^{ex} - \mu_1^{ex}) / k_B T} τ1→TS1∝e−(μTSex−μ1ex)/kBT
假设 μ T S e x − μ 1 e x ≫ k B T \mu_{TS}^{ex} - \mu_1^{ex} \gg k_B T μTSex−μ1ex≫kBT,则 μ 1 e x \mu_1^{ex} μ1ex表示在温度 T T T下长寿命的稳定态。
向前跃迁的净平均速率为:
R 1 → 2 = R 0 e − ( μ T S e x − μ 1 e x ) / k B T R_{1\to 2} = R_0 e^{- (\mu_{TS}^{ex} - \mu_1^{ex}) / k_B T} R1→2=R0e−(μTSex−μ1ex)/kBT
因此,净反应速率为:
R = R 1 → 2 − R 2 → 1 R = R_{1\to 2} - R_{2\to 1} R=R1→2−R2→1
= R 0 [ e − ( μ T S e x − μ 1 e x ) / k B T − e − ( μ T S e x − μ 2 e x ) / k B T ] = R_0 \left[ e^{- (\mu_{TS}^{ex} - \mu_1^{ex}) / k_B T} - e^{- (\mu_{TS}^{ex} - \mu_2^{ex}) / k_B T} \right] =R0[e−(μTSex−μ1ex)/kBT−e−(μTSex−μ2ex)/kBT]
在热平衡时, μ 1 = μ 2 \mu_1 = \mu_2 μ1=μ2,且净反应速率 R = 0 R=0 R=0,如果我们正确定义了 μ1 和 μ2,则 R 1 → 2 0 = R 2 → 1 0 \mathcal{R}_{1\to2}^0=\mathcal{R}_{2\to1}^0 R1→20=R2→10 。
因此,我们得到了浓溶液中的反应速率的通用表达式:
R = R 0 [ e − ( μ T S e x − μ 1 e x ) / k B T − e − ( μ T S e x − μ 2 e x ) / k B T ] R = R_0 \left[ e^{- (\mu_{TS}^{ex} - \mu_1^{ex}) / k_B T} - e^{- (\mu_{TS}^{ex} - \mu_2^{ex}) / k_B T} \right] R=R0[e−(μTSex−μ1ex)/kBT−e−(μTSex−μ2ex)/kBT]
2. 能斯特方程
2.1 详细平衡
当两种准平衡态之间的平均跃迁速率相等时,系统处于平衡状态,这称为“详细平衡”。在详细平衡条件下,我们有:
μ 1 = μ 2 \mu_1 = \mu_2 μ1=μ2
详细平衡使得净反应速率 R = 0 R=0 R=0,从而可以推导浓溶液中的能斯特方程。
我们还有 de Donder relation:
R 1 → 2 R 2 → 1 = e ( μ 1 − μ 2 ) k B T \frac{\mathcal{R}_{1\to2}}{\mathcal{R}_{2\to1}}=e^{\frac{(\mu_1-\mu_2)}{k_BT}} R2→1R1→2=ekBT(μ1−μ2)
它将反应动力学(左侧)与平衡热力学(右侧)联系起来。
2.2 静电部分的分离
考虑一般的法拉第半电池反应:
∑ s i M i z i → n e − \sum s_i M_i^{z_i} \to ne^- ∑siMizi→ne−
我们可以将其表示为:
R → n e − + O R \to ne^- + O R→ne−+O
其中反应物和产物分别来自状态1和状态2。 R = ∑ i s i R M i R z i R a n d O = ∑ i s i O M i O z i O . R=\sum_{i}s_{iR}M_{iR}^{z_{iR}}\mathrm{~and~}O=\sum_{i}s_{iO}M_{iO}^{z_{iO}}. R=∑isiRMiRziR and O=∑isiOMiOziO.
化学势的静电部分可以分离表示为:
μ 1 = μ R + q R ϕ \mu_1 = \mu_R + q_R \phi μ1=μR+qRϕ
其中 μ R = k B T ln a R \mu_R = k_B T \ln a_R μR=kBTlnaR表示化学部分, q R = ∑ s i R z i R e q_R = \sum s_i^R z_i^R e qR=∑siRziRe为静电部分, ϕ \phi ϕ表示溶液电位。同理,
μ 2 = μ O + q O ϕ − n e ϕ e \mu_2 = \mu_O + q_O \phi - ne \phi_e μ2=μO+qOϕ−neϕe
其中, μ O = k B T ln a O \mu_O = k_B T \ln a_O μO=kBTlnaO, q O = ∑ s i O z i O e q_O = \sum s_i^O z_i^O e qO=∑siOziOe
在平衡条件下,de Donder 关系意味着:
1 = R 1 → 2 R 2 → 1 = e ( μ 1 − μ 2 ) / k B T = a R a O e q R ϕ − q O ϕ + n e ϕ e k B T . \begin{aligned} \text{1}& =\frac{\mathcal{R}_{1\to2}}{\mathcal{R}_{2\to1}} \\ &=e^{(\mu_1-\mu_2)/k_BT} \\ &=\frac{a_R}{a_O}e^{\frac{q_R\phi-q_O\phi+ne\phi_e}{k_BT}}. \end{aligned} 1=R2→1R1→2=e(μ1−μ2)/kBT=aOaRekBTqRϕ−qOϕ+neϕe.
利用电荷守恒 q R = q O − n e − q_R = q_O − ne^- qR=qO−ne−,可以得到浓溶液中的能斯特方程:
Δ ϕ e q = k B T n e ln a O a R \Delta \phi_{eq} = \frac{k_B T}{ne} \ln \frac{a_O}{a_R} Δϕeq=nekBTlnaRaO
我们可以写为:
Δ
ϕ
e
q
=
V
0
−
k
B
T
n
e
ln
a
R
/
a
R
0
a
O
/
a
O
0
\Delta\phi_{eq}=V^0-\frac{k_BT}{ne}\ln\frac{a_R/a_R^0}{a_O/a_O^0}
Δϕeq=V0−nekBTlnaO/aO0aR/aR0
其中
V
0
=
k
B
T
n
e
ln
a
R
0
a
O
0
V^0=\frac{k_BT}{ne}\ln\frac{a_R^0}{a_O^0}
V0=nekBTlnaO0aR0,这里
a
O
0
a_O^0
aO0和
a
R
0
a_R^0
aR0代表参考状态。
3. Butler-Volmer 方程
3.1 过渡态的假设
为了描述系统的反应速率及其动力学行为(非平衡状态下),我们需要一个过渡态的模型。我们采用Butler-Volmer假设,假设过渡态的静电能为:
μ T S e x = μ A + α q A ϕ + ( 1 − α ) [ q 0 ϕ − n e ϕ e ] \mu_{TS}^{ex} = \mu_A + \alpha q_A \phi + (1 - \alpha) [q_0 \phi - ne \phi_e] μTSex=μA+αqAϕ+(1−α)[q0ϕ−neϕe]
其中 μ A = k B T ln γ A \mu_A = k_B T \ln \gamma_A μA=kBTlnγA表示过渡态的超额化学活度,第二项表示过渡态的静电能,为状态1和状态2贡献的加权平均。
3.2 活化过电位的影响
通过类似的推导,我们可以将过渡态的化学势 μ T S e x \mu_{TS}^{ex} μTSex、状态1和状态2的化学势 μ 1 \mu_1 μ1和 μ 2 \mu_2 μ2代入到反应速率 R R R中,并通过平衡电位 Δ ϕ e q \Delta \phi_{eq} Δϕeq表示反应速率与活化过电位 η a c t = Δ ϕ − Δ ϕ e q \eta_{act} = \Delta \phi - \Delta \phi_{eq} ηact=Δϕ−Δϕeq的关系:
R = R 0 ( a R α a O 1 − α ) [ e ( 1 − α ) n e η / k B T − e − α n e η / k B T ] R = R_0 \left( a_R^{\alpha} a_O^{1-\alpha} \right) \left[ e^{(1-\alpha)ne\eta / k_B T} - e^{-\alpha ne\eta / k_B T} \right] R=R0(aRαaO1−α)[e(1−α)neη/kBT−e−αneη/kBT]
我们定义电流 I = n e A R I = ne A R I=neAR,其中 A A A为电极面积,得到:
I = I 0 [ e ( 1 − α ) n e η / k B T − e − α n e η / k B T ] I = I_0 \left[ e^{(1-\alpha)ne\eta / k_B T} - e^{-\alpha ne\eta / k_B T} \right] I=I0[e(1−α)neη/kBT−e−αneη/kBT]
其中交换电流 I 0 I_0 I0为:
I 0 = n e A R 0 ( a R α a O 1 − α γ A ) I_0 = ne A R_0 \left( \frac{a_R^{\alpha} a_O^{1-\alpha}}{\gamma_A} \right) I0=neAR0(γAaRαaO1−α)
交换电流 I 0 I_0 I0与反应物和产物的活度及活度系数有关,是反应速率的重要参数。
4. 总结
- 浓溶液中的反应速率需要考虑超额化学势,通过活动度来替代浓度,以更准确地描述反应动力学。
- 超额化学势包含了浓溶液中的相互作用,例如排斥体积效应和外部系统的贡献。
- 能斯特方程在浓溶液中可通过详细平衡推导得到,描述了电极电位与化学活动度之间的关系。
- Butler-Volmer方程描述了电极反应的动力学行为,考虑了过渡态的静电能和活化过电位。
- 交换电流与活度和活度系数有关,是影响反应速率的重要参数,其大小直接反映了反应的可逆性和动力学活性。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









