




本文来自“生信算法”公众号。
基因变异普遍存在于同一物种内的不同个体中,如人与人之间的基因组是不完全一样的(即是多态的),彼此之间都存在着一些差异,即使是和父母或是兄弟姐妹之间去比较。这种差异也是基因组多态性的来源,通过外在和内在特征表现出来,比如头发和眼睛颜色,高矮胖瘦,抵抗力等。这些差异也是造成我们彼此之间不同的一个重要原因。
基因变异是一个相对的概念,只有在彼此的比较中才有存在意义。基因变异主要分为三大类:
-
单核苷酸变异,通常称为单核苷酸多态性(single nucleotide polymorphism),就是单个DNA碱基的不同,简称SNP;
-
小的Indel(Insertion 和 Deletion的简),指的是在基因组的某个位置上所发生的小片段序列的插入或者删除,长度通常在50 bp以下;
-
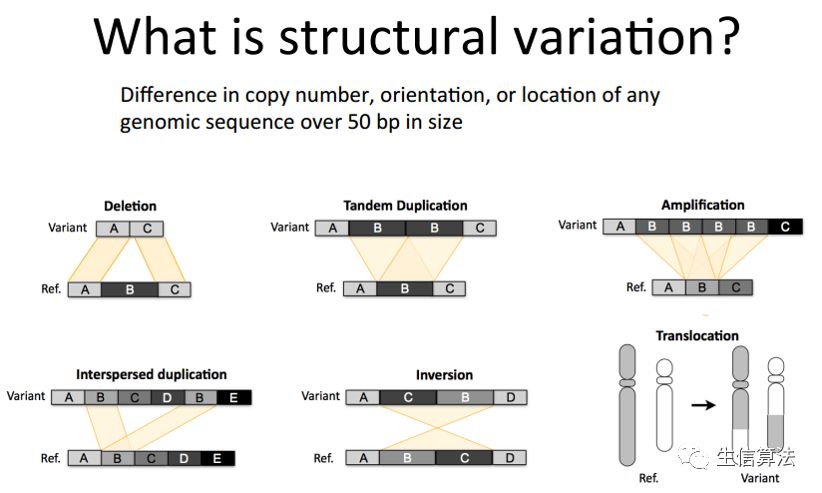
大的结构性变异,这种类型比较多,包括长度在50 bp以上的长片段序列的插入或者删除、染色体倒位,染色体内部或染色体之间的序列易位,拷贝数变异,以及一些形式更为复杂的变异。
为了和SNP变异作区分,第2和第3类变异通常也被称为基因组结构性变异(Structural variation,简称SV)。
结构变异长度少则几十bp,多则几万bp,对其检测带来了极大挑战。研究人员发现SV对基因组的影响比起SNP来说还要大,而且基因组上的SVs比起SNP而言,似乎更能用于解释人类群体多样性的特征。再者,一些SVs往往和一些疾病(包括癌症、自闭症、老年痴呆症)的发生相关联,甚至还是其致病的诱因。因此对SVs进行检测具有重要的意义。
二代测序技术产生的序列虽然精度高,但读段短(100~500 bp),不能跨越大多数长度较长的SVs区域,但基于单分子测序的三代测序技术(如PacBio,牛津纳米孔测序)可以测得长达100 kpb的序列,轻松跨越各种SVs区域,为SVs检测带来方便。但其错误率较高(~15%),因此需要专门的SVs检测算法。PBHoney是较早利用三代序列进行SVs检测的软件。
PBHoney方法
PBHoney主要利用两种序列比对情况进行检测。如下图所示,一种是Interrupted long-read mapping,一种是 Intra-read discordance。主要原因在于对于一条比对的三代序列,比对情况可以分为这两种。
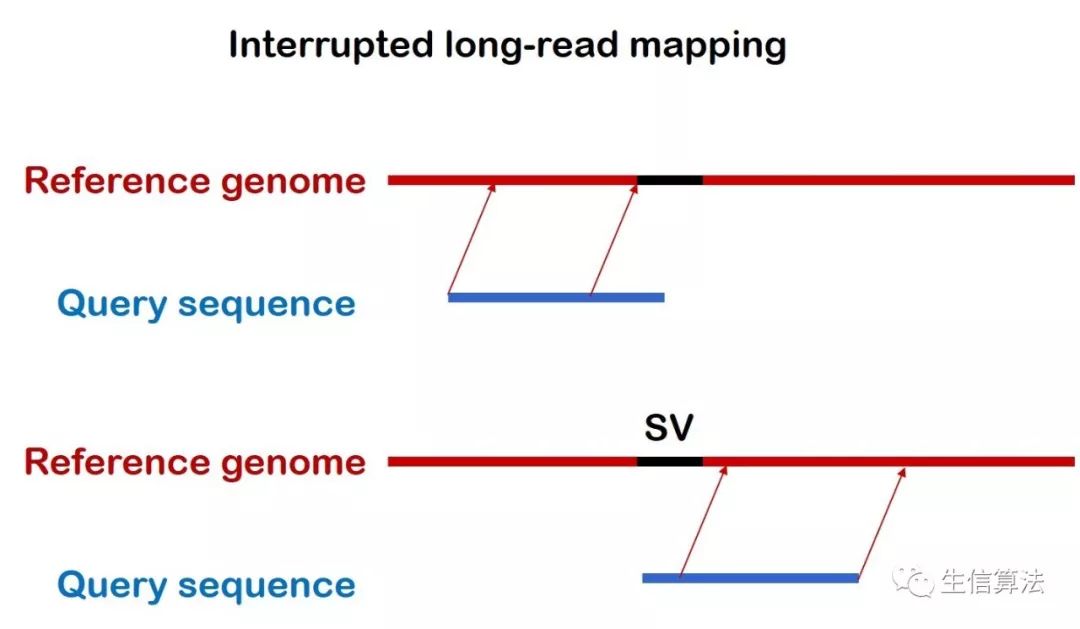
所谓Interrupted long-read mapping ,就是这条序列只比对了一部分,其前缀或者后缀的序列片段没有比对上去。
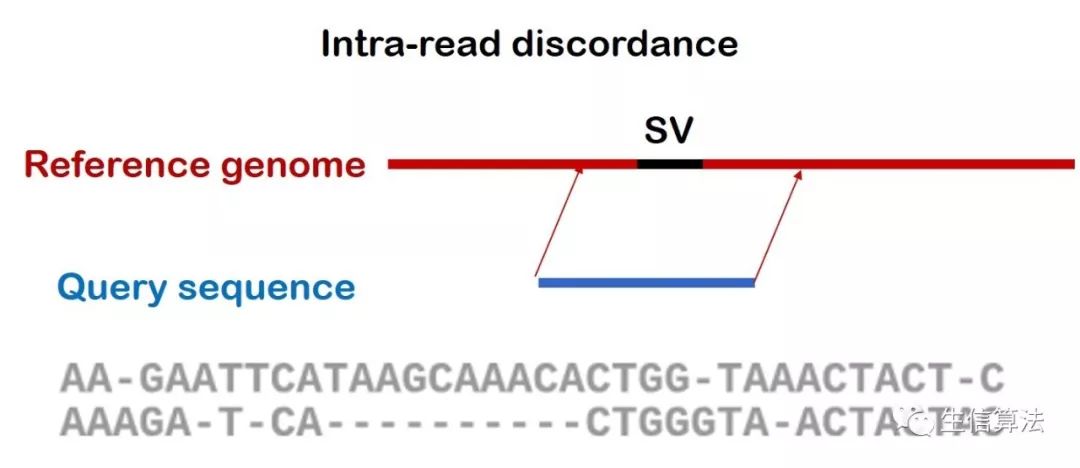
下图是 Intra-read discordance比对示意图,所谓 Intra-read discordance,就是整条序列都比对到参考基因组上,但某些比对区域由于SVs存在比对质量较低,存在较多非匹配状态。
PBHoney首先采用三代序列比对算法BLASR进行序列比对,然后针对这两种比对情况做相应的处理,找出SVs区域。
处理 Interrupted long-read mapping序列
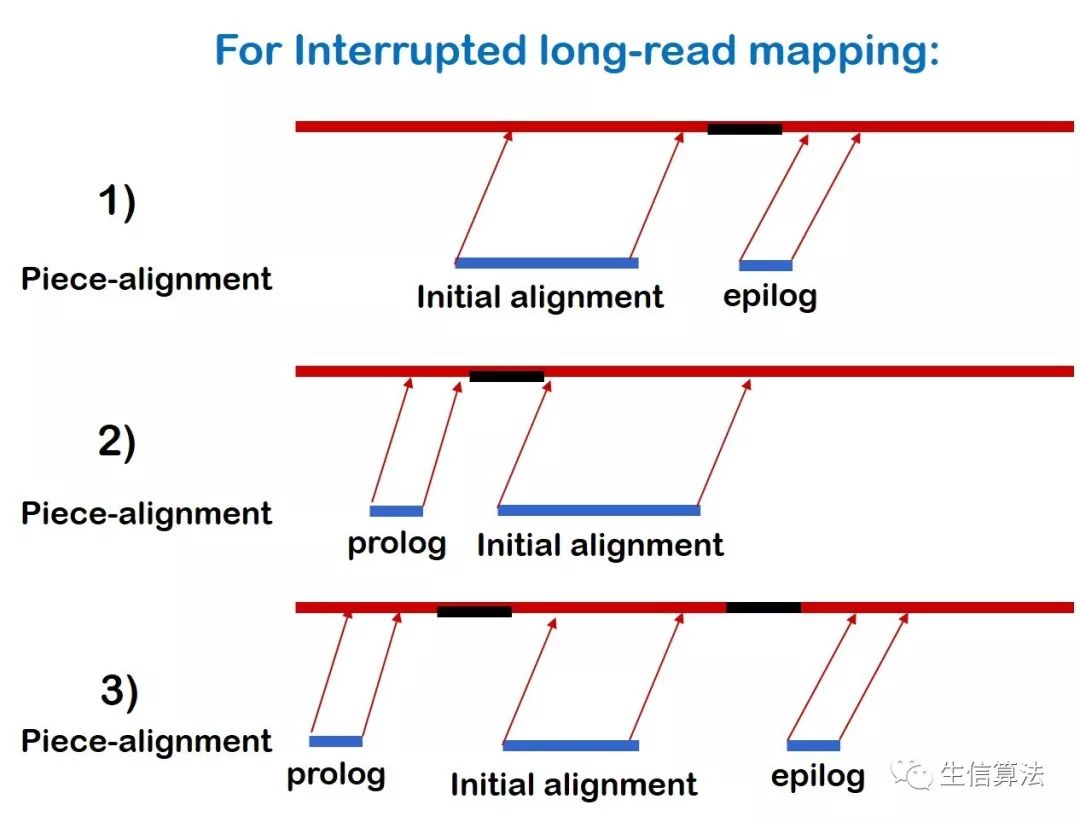
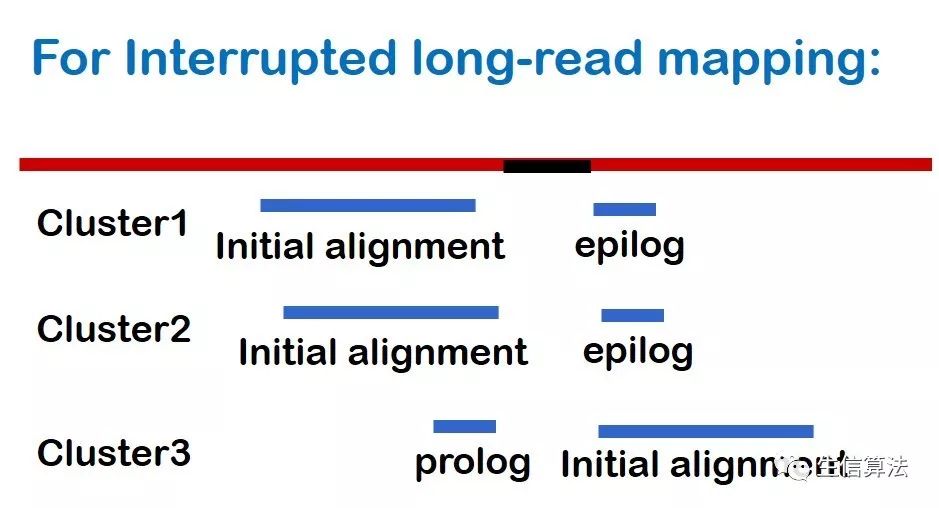
首先将提取没有比对的前缀或者后缀,如果长度大于200 bp,再将这些片段通过BLASR比对到参考基因组上。然后与原来比对的序列(initial alignment)组成piece-alignment,如下图所示。后缀比对的片段称为epilog,前缀比对的片段称为prolog。
然后对每个piece-alignment进行聚类处理:
- 只考虑包含initial alignment 和 epilog (或prolog)的piece-alignment。如果epilog和prolog都存在,(如上图3)所示,只考虑比对质量高的。
- 保留initial alignment 和 epilog (或prolog)之间的距离小于200 bp的piece-alignment。200 bp是默认值,用户可自己设置。保留下来的piece-alignment称为一个cluster。
- SVs的位置(起始位置和结束位置)就是每个cluster中比对位置的平均值。如下图所示。
处理 Intra-read discordance 序列
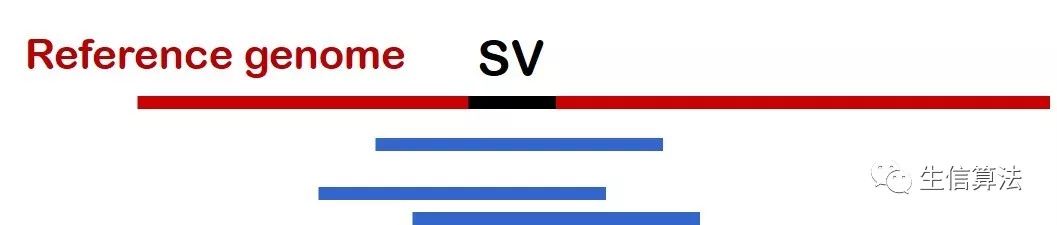
对于完整比对上的序列,如上图所示,通过以下步骤进行处理:
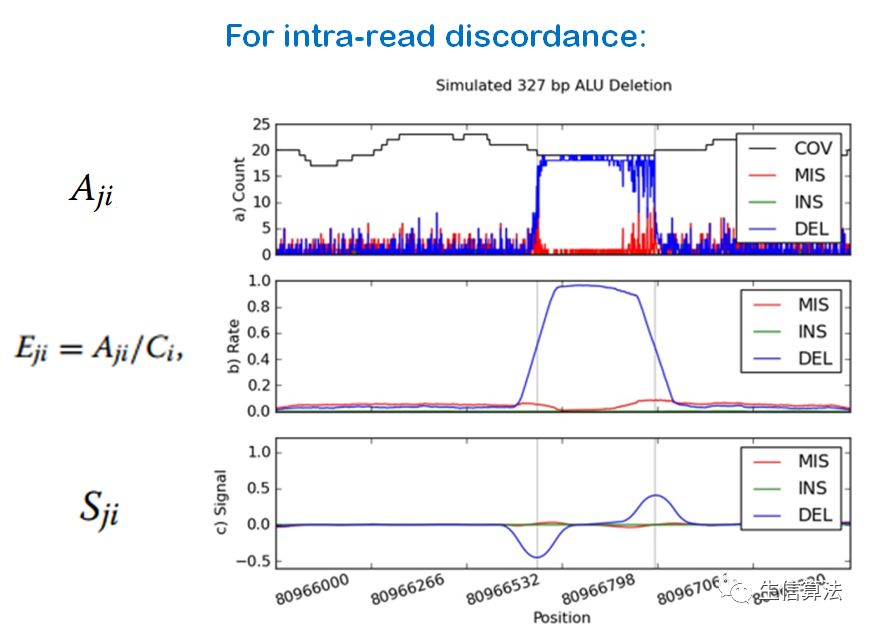
1. 计算比对到基因组序列每个位置的替换(mismatch)、插入、删除碱基个数、及覆盖率。构成一个4×G大小的数组A,其中G是参考基因组的序列长度。
2. 然后计算基因组每个位置的错误率大小:
其中j=1,2,3,4. Ci表示覆盖率。
3. 通过下面公式对E矩阵进行变换:
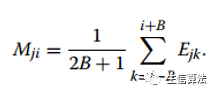
4. 再次对M矩阵进行变换:
其中步骤3)和4)目的是寻找一些差异比较大的值,更好判断是否是SV。
下图是通过以上步骤检测一个删除SV的示意图。可以看出步骤3)和4)可以将A柱状图转变成脉冲信号,方便SV检测。
结果
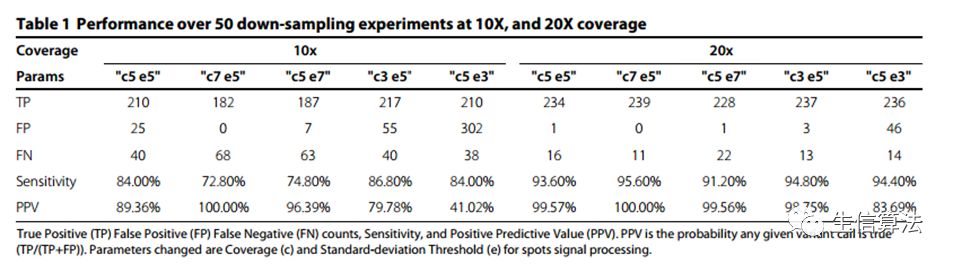
下表是PBHoney的检测结果,可以看出其灵敏度和PPV值还是不错的,这篇文章是以软件类(software)形式发表的,没有和别的方法进行比较。
总结
SVs检测对于疾病治疗具有重要意义,随着三代测序技术的不断发展,其测序准确性和序列长度会不断改善,相信基于三代序列的结构变异检测会得到越来越多的关注。
参考文献
- English A C, Salerno W J, Reid J G. PBHoney: identifying genomic variants via long-read discordance and interrupted mapping. BMC bioinformatics, 2014, 15(1): 180.
- https://blog.youkuaiyun.com/alex6plus7/article/details/50236375

















 682
682

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









