




本文来自“生信算法”公众号。
DNA和RNA是生命体的两种基本遗传物质,蕴含全部的遗传信息,测序技术能够真正揭示基因组的复杂性和多样性,为后续分析提供重要技术支撑。从1952年科学家确定DNA为遗传物质,到1966年全部密码子被破解后,在基础生物学研究与应用领域,获取DNA序列已成为不可缺少的知识。
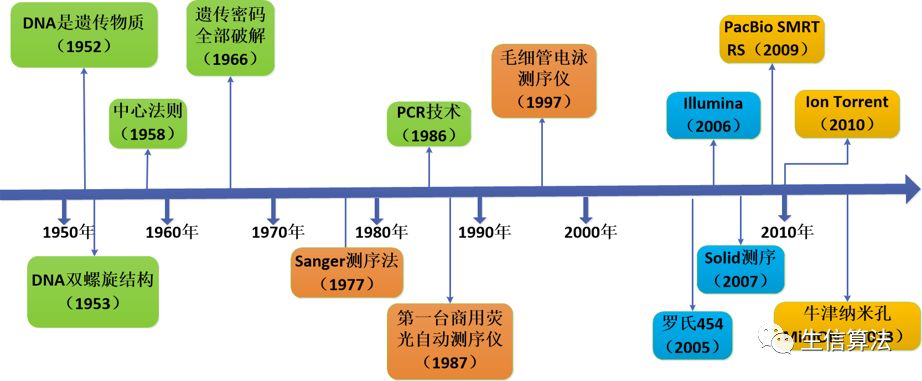
在这种迫切需求下,1977年以sanger测序法为代表的第一代测序技术应运而生,并被不断改进。直至2005年以罗氏454为代表的第二代测序技术的出现,开启了高通量测序时代,在二代测序早期,454,illumina与SoLid呈三足鼎立之势。随着时间的推进以及测序技术的不断创新,以PacBio为代表的第三代测序技术横空出世,随后也出现了IonTorrent 与牛津纳米技术测序,推动着测序技术的不断发展。
01 Sanger测序法

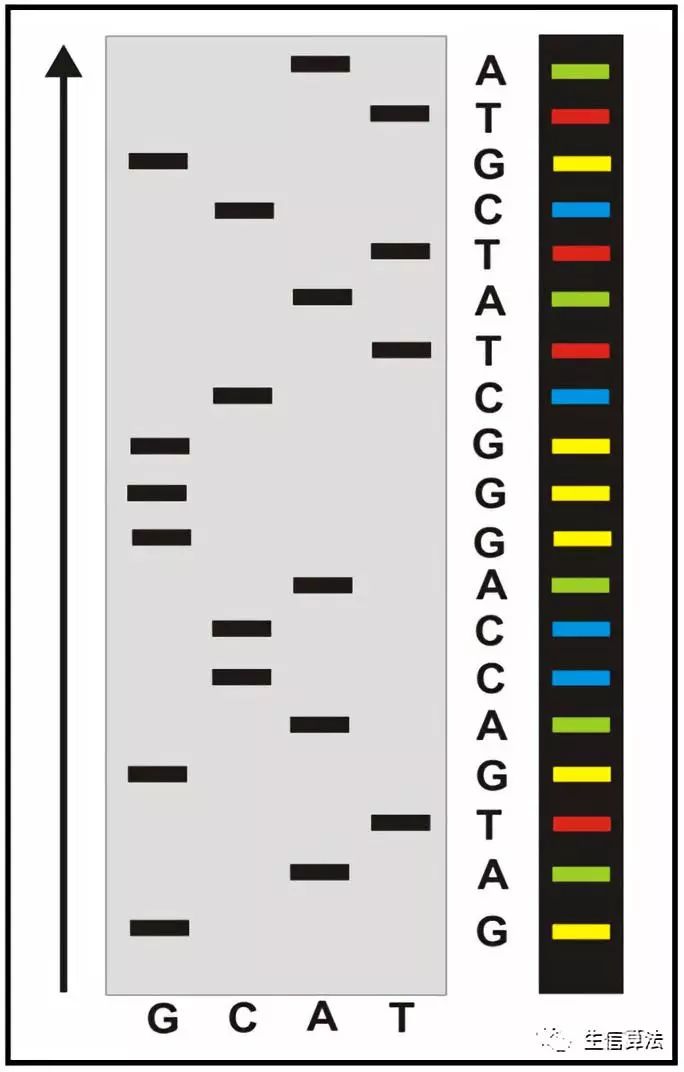
早期的DNA测序主要通过Sanger测序技术测定,该技术是上世纪七十年代由Sanger建立的DNA测序法,使读取DNA碱基序列成为可能。该方法首先采用PCR扩增策略获得待测核算模板,然后在四支试管中加入适当引物、模板、dNTP及ddNTP进行延伸,由于ddNTP在不同位置参入,因而产生了一系列不同长度的新DNA链,最后分四个泳道进行电泳分离便可依次阅读合成最终碱基序列。
02 二代测序技术
由于Sanger测序技术依赖电泳分离技术,在分析速度和并行化方面很难有进一步提升,其测序成本也很高,尤其是进入二十一世纪后,随着实验需求的增长及效率要求的提高,需要寻求新的测序方法突破这些局限。在这一背景下,新一代测序技术又称二代次序技术应运而生,早期主要包括罗氏454公司的GS FLX测序平台、Illumina公司的Solexa Genome Analyzer测序平台和ABI公司的SOLiD测序平台。在二代测序技术出现早期,三家测序技术呈现三足鼎立之势。随后Illumina凭借其通量高,成本低,测序速度快等优势垄断了全球测序仪器市场超过70%的份额,不断推出新的测序仪(如MiSeq,HiSeq,NextSeq等)。这家全世界最大的DNA测序公司早已不在局限于向用户销售设备,通过一系列收购、战略合作等手段,Illumina业务范围早已渗透测序行业上、中、下游,连续多年被MIT评为“世界最聪明公司”。

03 三代测序技术
随着二代测序技术的不断发展,其以更高的通量以及不断降低的测序费用加速拓展着在测序研究领域的广度和深度,但其不足之处也日益凸显。例如,在微生物分析领域,由于该技术原理是建立在PCR基础之上,扩增后得到的DNA分子片段的数目和扩增前DNA分子片段的数目比例有相对偏差,而且具有GC偏好性,对物种多样性分析有较大影响,尤其是对于少量物种影响更大。尽管Illumina的测序仪器不断更新,但这些缺点是其固有的,并没有被克服。更大的不足之处是二代测序技术读长较短(~300bp),对后续的序列拼接,组装以及跨越重复区域等生物信息学分析带来较大困难。所以迫切需要技术革新增加测序长度,在这种情况下,第三代单分子测序技术应运而生。
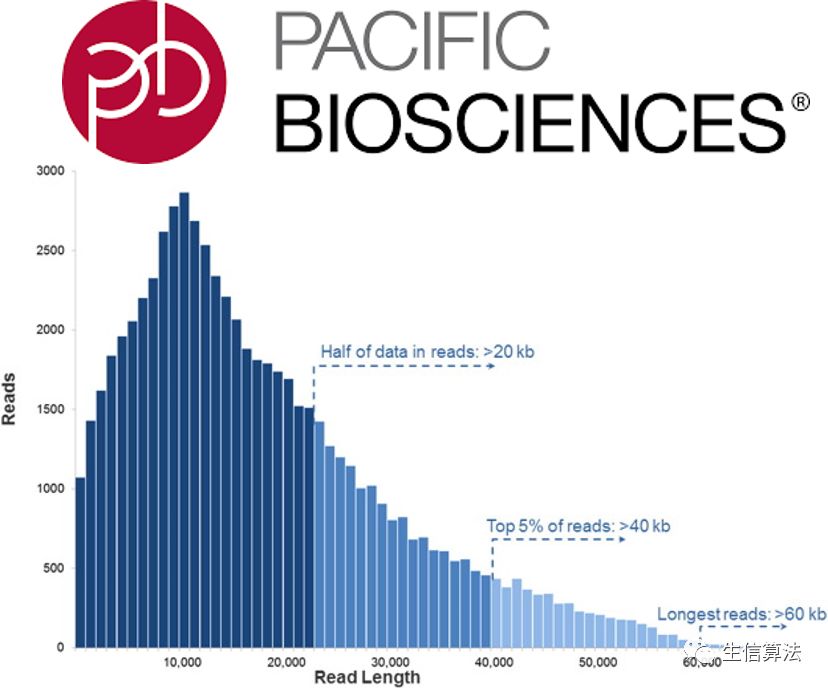
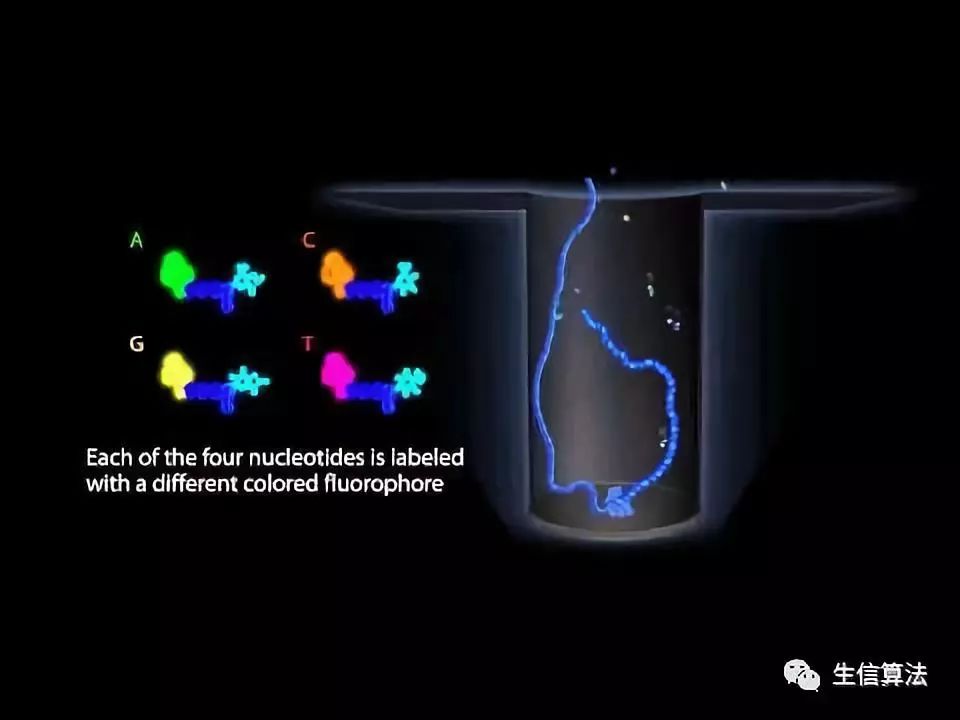
在第三代测序公司中,PacBio(Pacific Biosciences)公司推出的单分子实时测序系统PacBio RS,首次实现了对单个DNA分子合成的实时观测,也是首个将其成功商业化的公司。PacBio从第一代的PacBio RS测序仪,到最新的Sequel测序仪,都采用独特的SMRT Cell专利技术,每个Cell有150万个纳米级的零模波导孔(zero-mode waveguides,ZMWs),每个ZMW都能够包含一个DNA聚合酶及一条DNA样品链,测序反应在其中进行。首先最大的优势是长读长,平均读长可以达到8kbp,是Illumina测序平台的20-30倍。其次,PacBio SMRT采用单分子实时检测方式,模板制备时无需PCR扩增,每一条模板链都可以获得相应的序列信息,排除了PCR过程中有可能产生的错误,也排除了其他分子的干扰;覆盖深度也不受序列中GC含量差异影响,数据覆盖度更加均匀准确。最后PacBio的样品制备简便快捷,仪器操作简单,从样本到获取序列信息可在一天内完成。所以,PacBio有着二代测序技术无法比拟的优势。
04 BLASR比对工具
面对这些读段长,错误率高(~15%)的三代序列,需要开发专门的序列比对软件。BLASR是第一个针对PacBio序列的比对工具,2012年发表在《BMC Bioinformatics》期刊上,由PacBio研究团队开发,并且一直在更新,目前Google引用次数为433(截止2018.08.14)。其主要思想如下图所示:
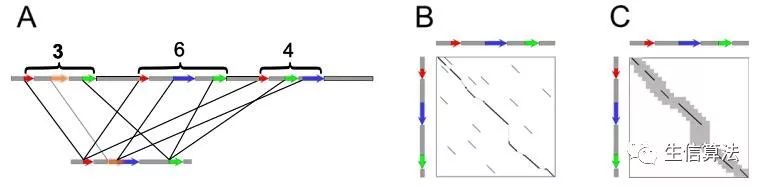
包含三步:
- 首先通过BWT-FM压缩或后缀数组(suffix array)索引技术对基因组进行转换,寻找与待比对序列相似性比较高的候选区域或区间;
- 然后对这些候选区间进行稀疏动态比对(sparse dynamic programming)得到初步比对结果;
- 最后运用动态规划算法进行详细的序列比对,得到最终的比对结果。
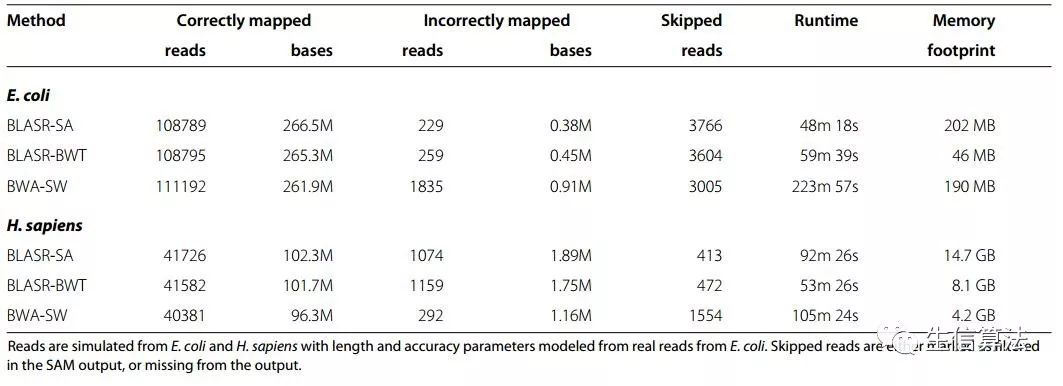
由于BLASR运用BWT-FM索引技术,因而可以大大提高搜索速度,并且降低内存消耗。下图是BLASR与传统二代比对工具的运行比较结果。可以看出不论运行速度方面还是消耗内存方面,BLASR均优于BWA-SW算法。
05 BLASR安装运行
BLASR安装非常简单,博哥已将其安装在自己笔记本电脑上,并测试成功,按照下面方法一步一步来即可。
(1)安装Biocoda或Minicoda。
Conda 是一个开源的软件包管理系统和环境管理系统,用于安装多个版本的软件包及其依赖关系,并在它们之间轻松切换。而Bioconda是Conda专门为生信开的一条通道,里面有非常多的软件,可以直接安装。下面介绍MiniCoda的安装,在Coda网站上(https://conda.io/miniconda.html)下载适合自己的版本,博哥笔记本是Linux 64位系统,直接下载Minicoda64位版本。
下载好后直接运行,一直按照提示进行安装:
./Miniconda2-latest-Linux-x86_64.sh
(2)安装BLASR
重新打开终端,直接输入并运行:
conda install -c bioconda blasr
然后会自动安装,直至安装完毕。如果中间网速慢断开,可重复运行安装。

(3)运行BLASR
终端直接输入:
blasr sequence.fasta -m 0 --bestn 1 --nproc 16 >blasr_align.txt
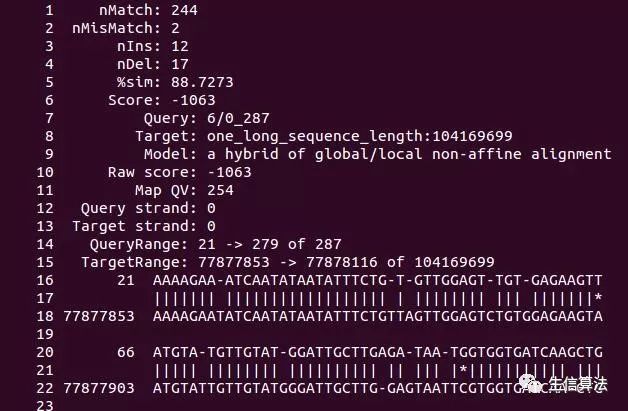
其中sequence.fa是带比对序列的FASTA格式(FASTQ也可以),sequence.fa是基因组序列,-m 0 指的是输出类似于BLAST的比对格式,见下图。--bestn 1指的是只输出1条最相似的比对结果,--nproc是线程数。>blasr_align.txt是输出结果。BLASR比对结果如下:

今天的介绍到此结束,感谢大家的阅读和关注,欢迎大家留言~
参考文献:
Chaisson M J, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory[J]. BMC bioinformatics, 2012, 13(1): 238.

关注生信算法|掌握算法奥秘


















 4737
4737

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









