




可变剪切因子 motif 分析
可变剪切调控因子与转录因子类似,具有特定的 RNA 结合 motif,是一种 RNA-binding protein。
-
相同点:
-
都是蛋白质的序列结合区域
-
有特定的序列 motif
-
-
不同点:
-
TF 的 motif 主要结合在 promoter 和 enhancer,负责基因转录
-
ASF 的 motif 主要结合在 gene 的 intro 区域,负责可变剪切
-
(一)获取可变剪切因子 motif
- 直接从 MEME 数据库中获取 motif 文件
MEME 工具含有多个 motif 数据库,具体可用数据库可参看:https://meme-suite.org/meme/db/motifs

所有 motif 数据库可直接下载使用:https://meme-suite.org/meme/doc/download.html
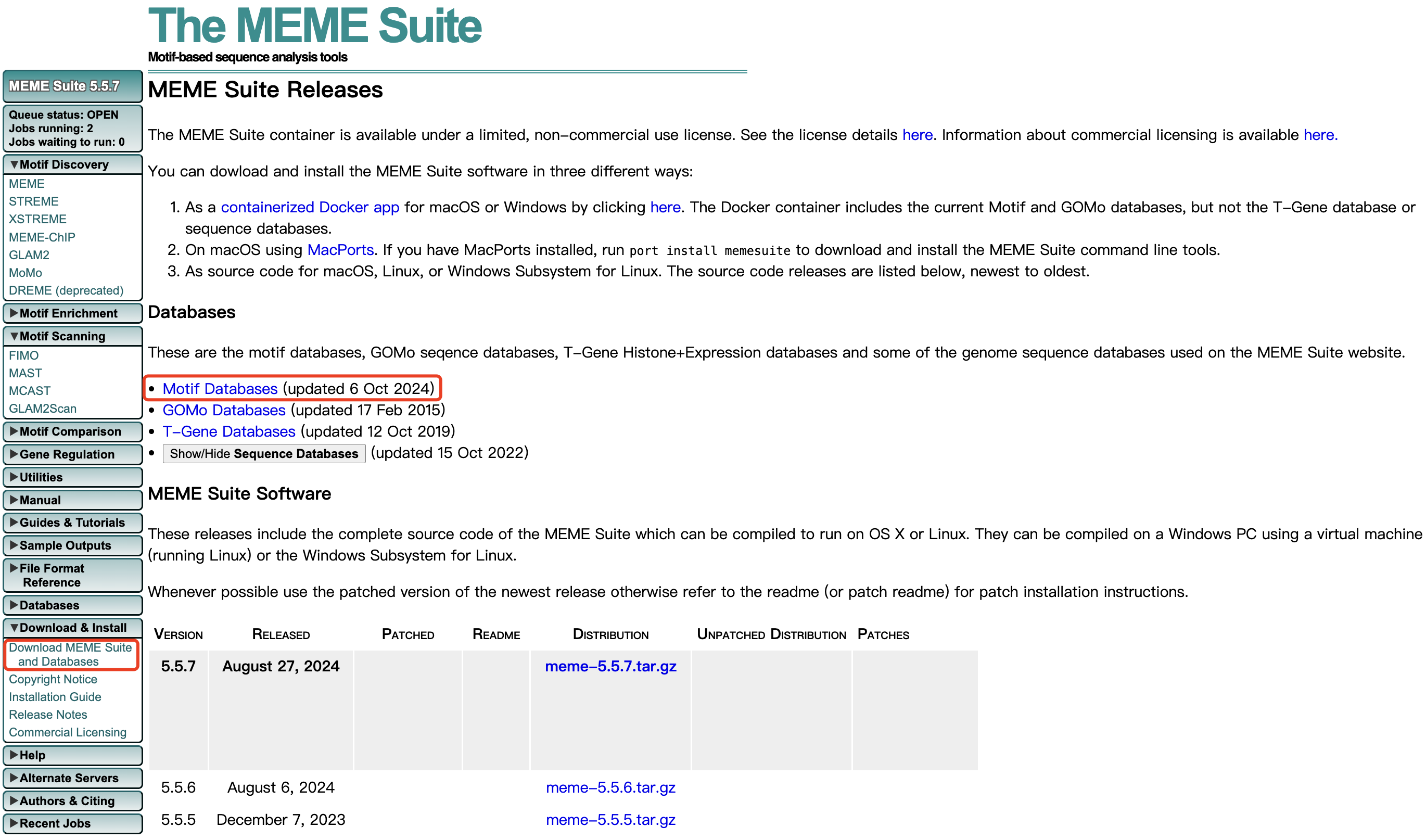
在 motif 数据库详情页面可查看下载压缩包中具体文件所在路径:
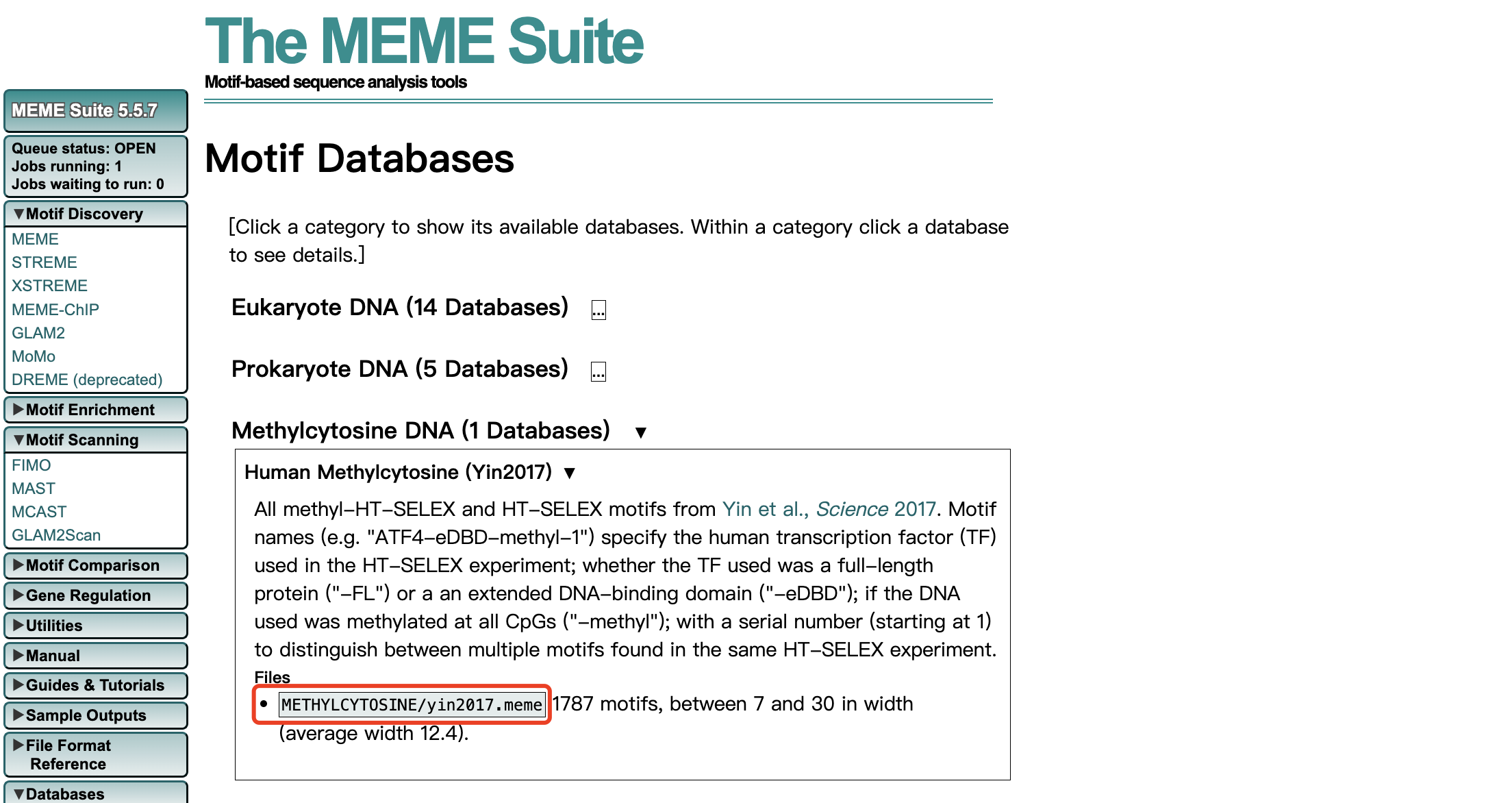
此处使用的 PTBP1 示例文件可以直接在 MEME 下载 CISBP-RNA 数据库(http://cisbp-rna.ccbr.utoronto.ca/)信息,并从中提取对应剪切因子的 motif,其中 MEME 中提供了 CISBP-RNA 数据库的 RNA motif 和 DNA motif 。
MEME version 5.5.5 (Thu Sep 14 08:48:04 2023 +1000)
ALPHABET= ACGU
Background letter frequencies (from uniform background):
A 0.25000 C 0.25000 G 0.25000 U 0.25000
MOTIF M227_0.6 PTBP1
letter-probability 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 3191
3191

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









