




文章目录
IGV——基因组可视化(二)
本部分为基于生信修炼手册的《玩转基因组浏览器》系列的笔记大汇总,对于初级的 IGV 使用进行了更为高阶的补充。
常见的基因组浏览器包括 UCSC Genome Browser、Ensembl Genome Browser、JBrowse 和 IGV,其中前三者都是基于网页,而 IGV 不仅提供了 web 服务,还提供了独立的安装包,允许在本地运行,同时还提供了命令行版本的运行工具 igvtools。
IGV 支持芯片数据,NGS 数据,基因组注释等多种类型的数据,各种类型的信息以行为单位进行展示,称为 track,不同类型的数据可视化方式不同。
官网:https://software.broadinstitute.org/software/igv/
网页版:https://igv.org/app/
(一)参考基因组
1. 下载内置参考基因组
IGV 软件内置有多个物种的参考基因组,可以直接通过 IGV 工具下载载入。
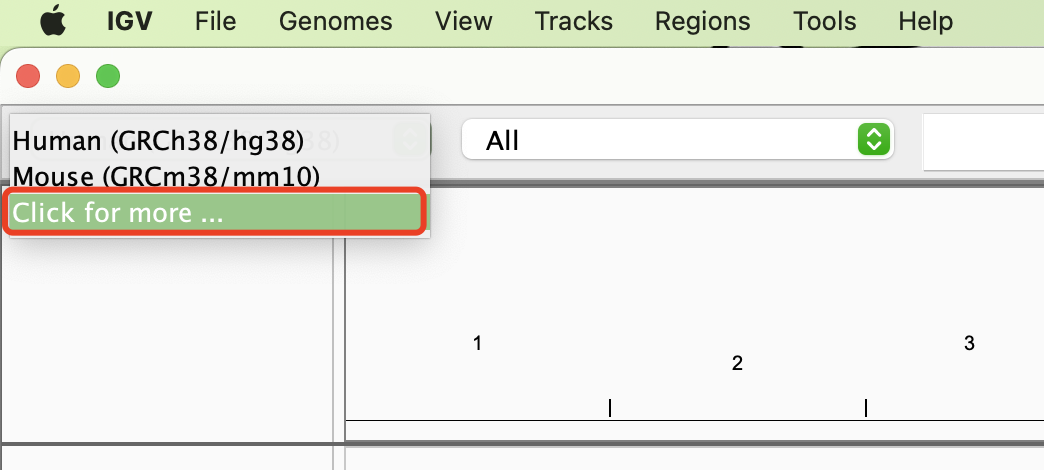
2. 自定义参考基因组
由于网络或其他原因,载入内置参考基因组时可能会失败,此时可以在本地构建参考基因组,使用时从本地导入即可,该方法即使在没有网络的情况下也可以使用。
构建本地参考基因组需要以下文件:
-
genome fasta:参考基因组的fasta文件,可以是一个文件包含了所有的染色体,也可以是一个目录,目录下每条染色体是一个单独的文件**(必须)**
-
cytoband file:染色体条带文件(可选)
-
gene annotation file:基因结构注释文件,支持bed、gtf、genePred 3种格式(可选)
-
alias file:别名,当fasta文件和基因结构中的染色体名称不同时,可以通过这个文件来进行映射(可选)
通常情况下,只需要基因组序列和基因结构文件即可满足需求。
注意:从版本 2.11.0 开始,通过“.genome” 格式指定参考基因组已被弃用,现在指定参考基因组为 JSON 文件(https://github.com/igvteam/igv/wiki/JSON-Genome-Format)。
(1)对于 fasta 文件,需要先建立后缀为 .fai 的索引。
# conda install -c bioconda samtools
samtools faidx hg19.fa
(2)根据文件 url 或路径创建基因组 JSON 文件。
必填字段为 “id”、“name”、“fastaURL” 和 “indexURL”,所有其他字段都是可选的。
URL 属性(所有以 url 结尾的字段)可以是绝对或相对文件路径,相对路径被解释为相对于基因组 json 文件的位置,如以下定义假定注释文件 gencode.v24.genes.gtf.gz 与 json 文件位于同一目录。
{
"id": "hg19",
"name": "Human (CRCh37/hg19)",
"fastaURL": "https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/hg19/hg19.fasta",
"indexURL": "https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/hg19/hg19.fasta.fai",
"cytobandURL": "https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/hg19/cytoBand 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 587
587

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









