



弗吉尼亚理工(Virginia Tech)的 Debswapna Bhattacharya 团队在《细胞系统》(Cell Systems)期刊上发表了一篇文章。

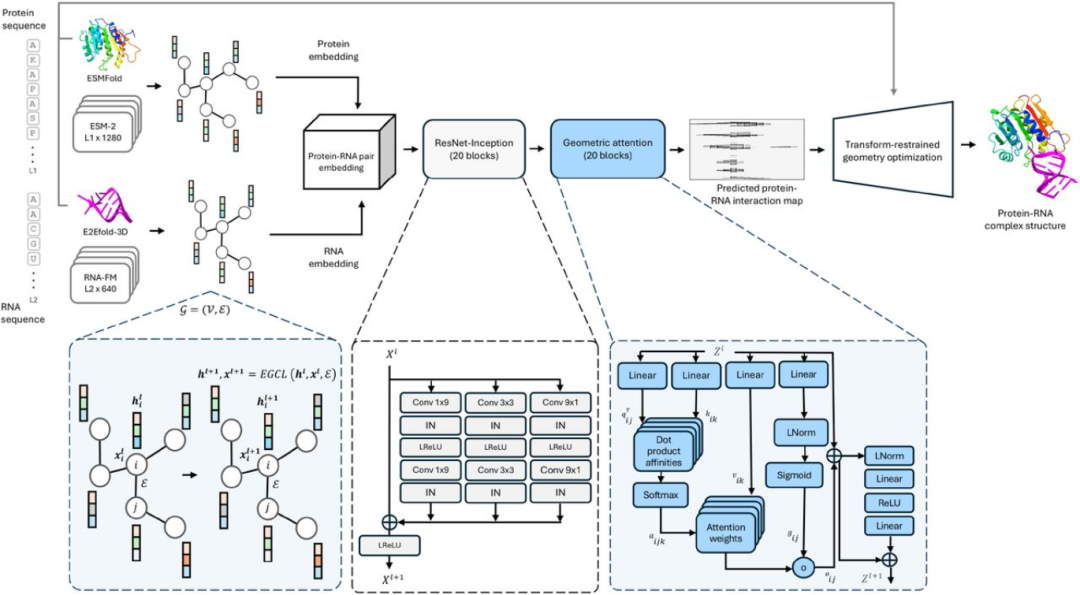
他们开源了一个名为ProRNA3D-single的AI工具。这东西只靠一条蛋白质序列和一条RNA序列,就能预测出它俩搅和在一起之后的三维立体结构。
在这个赛道上,即便是DeepMind大名鼎鼎的AlphaFold 3,表现也只能算差强人意。
AI预测界的旧王与新贵
生命活动的核心,很多时候就是蛋白质和RNA这对CP在干活,比如基因调控、病毒复制。搞清楚它俩是怎么抱在一起的,也就是解析出复合结构,就能从根本上理解很多疾病,开发新药。
但这件事,非常难。
传统的实验方法,比如X射线晶体学、冷冻电镜(cryo-EM)、核磁共振(NMR),要么样品难搞,要么太贵太慢,要么只能看小个头的分子。结果就是,在庞大的蛋白质结构数据库(PDB)里,带RNA的结构连2%都不到,缺口巨大。
于是,大家把希望寄托在计算方法上,尤其是AI。
AI界的王者AlphaFold 2在蛋白质结构预测上封了神,但它的升级版AlphaFold 3,在预测蛋白质-RNA复合物这个更复杂的任务上,却栽了跟头。根据公开数据,AlphaFold 3的测试集平均iLDDT(互作局部距离差异检验,可以理解为预测互作界面的准确分)只有39.4分。
另一个来自华盛顿大学贝克实验室的知名工具RoseTTAFold2NA(RF2NA),分数更惨,只有19.0。
这些工具都依赖一个叫多重序列比对(MSA)的东西。简单说,就是把一堆沾亲带故的序列放一起比对,从中寻找进化的蛛丝马迹来推断结构。可蛋白质和RNA的联合MSA数据,稀疏得可怜,噪声还大,严重拖了后腿。
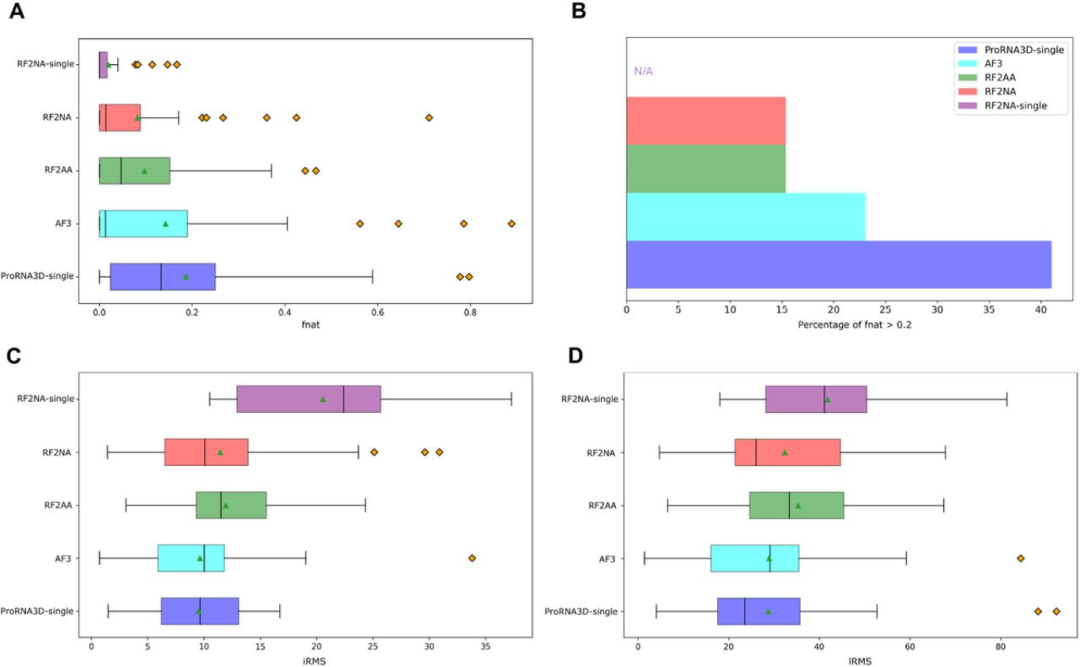
ProRNA3D-single的出现,就是来解决这个难题的。它不需要MSA,直接单挑,只用一条序列输入。结果显示,它的表现“令人信服地超越了包括AlphaFold 3在内的当前最先进方法,尤其是在进化信息有限的情况下”。
尤其是在进化信息稀缺的场景,比如预测新冠病毒(SARS-CoV-2)的RNA如何与人体蛋白勾结时,AlphaFold 3的准确率大幅下降,ProRNA3D-single却稳如泰山。
底气来自“生物语言模型”
ProRNA3D-single站在了巨人的肩膀上。
这个巨人,就是生物语言模型(bio LLMs)。你可以把它理解成生物界的ChatGPT,只不过它读的不是人类语言,而是蛋白质和RNA的序列“语言”。
ProRNA3D-single用了两个顶级的语言模型:
一个是Meta AI开发的ESM-2(第二代进化尺度模型),它“读”了2.5亿条蛋白质序列,对蛋白质的语言模式了如指掌。
另一个是北京大学和深圳湾实验室联合开发的RNA-FM(RNA基础模型),它“读”了上百万条RNA序列,是RNA界的语言大师。
当一条蛋白质和RNA序列被输入,这两个模型会分别输出一串数字,也就是“嵌入”。这串数字里,已经包含了序列自身隐含的进化和结构信息,从而摆脱了对外部MSA数据库的依赖。
有了高质量的嵌入,ProRNA3D-single开始了自己的骚操作,分三步走:
第一步,画图。它把蛋白质和RNA的嵌入信息,变成两张结构感知图。图里的每个点是一个氨基酸或核苷酸,点之间的连线代表它们的距离。
第二步,配对。这是最核心的创新。它用一个叫“配对模块”的东西,把两张图融合起来。这个模块里有个关键技术叫“几何注意力层”。普通的注意力机制只关心序列位置,而几何注意力机制能同时理解三维空间的几何约束,比如距离、角度、方向。它通过多头注意力机制,模拟多体互作,最终输出一张蛋白质-RNA的互作原子图谱,上面标明了哪里可能接触、距离多远、方向如何。
第三步,建模。有了这张精细的图谱,就等于有了施工蓝图。ProRNA3D-single把图谱信息转化成几何约束,然后用优化算法和分子动力学(MD)模拟,像搭积木一样,快速生成初始结构,再精细打磨,最终输出一个标准的PDB格式三维结构文件。
整个过程基于Python和PyTorch框架,在一块英伟达(NVIDIA)A100显卡上,预测一个中等大小的复合物(比如100个残基的蛋白+50个核苷酸的RNA),也就一个小时左右。
这玩意儿到底能干啥
ProRNA3D-single最直接的应用,是给RNA靶向药物的研发踩上一脚油门。
过去,药物研发主要盯着蛋白质靶点。现在,RNA靶向药成了新风口,它特异性高,副作用小。人类基因组里大概有1500个RNA结合蛋白(RBPs)和疾病有关,其中好几百个都被认为是“可成药”的靶点。但想设计药物,首先得知道靶点的三维结构,ProRNA3D-single正好能提供这个关键信息。
具体来说,有三大应用场景:
首先是抗病毒。病毒的复制、组装,处处离不开病毒RNA和宿主蛋白质的相互作用。比如新冠病毒的聚合酶就需要和人体蛋白结合才能干活。用ProRNA3D-single预测出这些结合界面,就能为设计药物、阻断病毒复制提供精确的“靶子”。
其次是神经退行性疾病。在阿尔茨海默病、渐冻症(ALS)里,像TDP-43这样的RNA结合蛋白会异常聚集,这是核心病理特征。ProRNA3D-single可以预测这些蛋白和RNA的正常及异常结合模式,帮助科学家理解病变是如何发生的。
再者是肿瘤治疗。很多癌症的发生,都和异常的RNA-蛋白互作有关,它们会激活癌基因,或者让抑癌基因沉默。比如MYC基因的信使RNA(mRNA)和一种叫IGF2BP1的蛋白结合后会变得更稳定,从而促进肿瘤生长。ProRNA3D-single可以帮助找到这些关键的互作,为开发靶向治疗提供思路。
弗吉尼亚理工官方新闻稿里,Bhattacharya教授说得很直白:“我们的终极目标是加速药物发现过程,阻止RNA病毒与宿主蛋白的相互作用,从而在疫情大流行之前阻止感染,或者抑制阿尔茨海默病中RNA结合蛋白的异常功能。”
开源、局限与未来
Bhattacharya团队遵循“开放科学”原则,把ProRNA3D-single的代码、预训练模型和文档全部放在了GitHub上开源。
当然,ProRNA3D-single也不是完美的。
团队在论文里坦诚,当前版本主要处理单链RNA,对双链RNA支持还不够;输出的是静态结构,没法模拟分子间相互作用的动态过程;也没考虑蛋白质翻译后修饰的影响。而且,对于特别巨大的复合物,预测精度也会下降。
这些也是他们未来的改进方向:支持多链RNA、结合分子动力学做动态建模、引入修饰信息、用分层策略搞定大分子机器。
数据方面,由于训练数据主要来自PDB数据库,存在一定偏差。比如,数据库里的物种主要是人、酵母、细菌,RNA类型也集中在核糖体RNA(rRNA)和转运RNA(tRNA)上,与疾病直接相关的复合物占比不到15%。团队已经采用了一些技术手段来缓解这些偏差。
从AlphaFold的横空出世,到ProRNA3D-single的精准打击,AI正在以一种超乎想象的速度,深入到生命科学最核心的领域。
AI已经开始预测生命的微观结构了,用AI优化甚至创造新物种将成为可能吗?
参考资料:
https://www.cell.com/cell-systems/abstract/S2405-4712(25)00233-9
https://www.sciencedirect.com/science/article/pii/S2405471225002339
https://github.com/facebookresearch/esm
https://academic.oup.com/nar/article/52/1/e3/7369930
https://github.com/Bhattacharya-Lab/ProRNA3D-single
https://www.biorxiv.org/content/10.1101/2024.07.27.605468v1.full-text
END

















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









