



真核生物基因组DNA以染色质形式存在,阐明其基因表达调控机制,关键在于解析染色质环境下蛋白质与DNA的相互作用。ChIP-seq技术整合了染色质免疫沉淀(ChIP)与新一代测序(NGS),可高效在全基因组范围内定位与组蛋白修饰、转录因子等蛋白结合的DNA区域。然而,从实验设计到数据产出,ChIP-seq研究中潜藏着诸多关键阶段,本文聚焦一些高频常见问题,系统性梳理从样本准备、实验设计到生信分析的完整链路,助力您的研究。
Q1:ChIP-seq的大概流程是怎样的?
(1)染色质交联与制备:使用1%甲醛处理细胞。甲醛介导的交联反应将蛋白质与DNA之间松散的物理相互作用(氢键)转化为稳定的共价键,形成牢固的蛋白-DNA复合物,防止后续操作中解离。交联后,裂解细胞膜释放内容物,并进一步分离纯化细胞核,从中提取交联染色质作为起始材料。
(2)染色质片段化:采用超声破碎通过高频声波产生的物理剪切力,将交联染色质随机打断成200-1000 bp(通常目标范围在 200-600 bp)的小片段。小片段化是免疫沉淀有效进行的前提,增大抗体可及性。另外也符合高通量测序平台对DNA片段长度的要求,并提高后续定位结合位点的分辨率。
(3)免疫沉淀IP:利用抗原-抗体特异性结合 富集目标蛋白及其结合DNA。可以直接使用针对目的蛋白 (如特定转录因子或组蛋白修饰标志) 的高质量、高特异性 ChIP级抗体。当缺乏优质内源抗体时,可对目标蛋白进行转基因加标签,使用相应标签抗体进行沉淀。将片段化染色质与特异性抗体孵育,形成抗体-蛋白-DNA复合物。加入Protein A/G磁珠。Protein A/G 能高亲和力结合抗体的 Fc 段,通过磁性分离将整个复合物(抗体-目标蛋白-结合DNA)沉淀/拉取下来。然后进行严格洗涤,去除非特异性结合的染色质片段。
(4)解交联、DNA纯化与生信分析:对沉淀得到的复合物进行解交联处理, 逆转甲醛交联,破坏共价键,释放出与目标蛋白结合的DNA片段。去除蛋白质等杂质,获得纯净的目标富集DNA片段。然后末端修复、加接头 、PCR 扩增后进行高通量测序和生信分析。对测序产生的海量短读段进行比对、峰识别(Peak Calling) 、注释、可视化等分析,最终在全基因组范围内定位目标蛋白的结合位点。也可以用获得的目标DNA片段,针对已知或候选的结合位点,使用特异性引物进行实时定量PCR,快速验证富集效果和特定区域的结合强度。
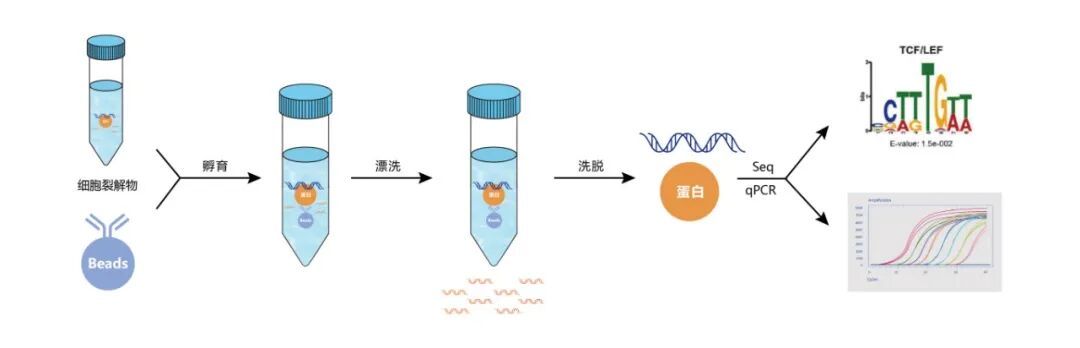
图 染色质免疫沉淀ChIP-seq/qPCR实验流程。
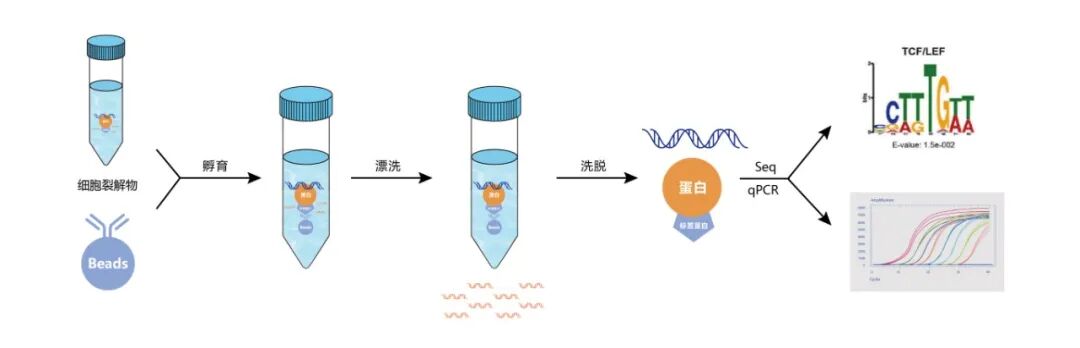
图 带标签的染色质免疫共沉淀流程图。
Q2:ChIP-seq单个样本包含的对照设置。
1、IP实验样本(核心实验组)
IP样本是通过抗体富集目标蛋白结合的染色质片段后获得的DNA。它是实验的核心,用于捕获特定蛋白质(如转录因子)结合的基因组区域。在测序中,IP样本的DNA占比会因抗体富集而升高,从而在后续分析中形成可检测的峰(peak)。
2、自身对照Input样本(关键背景参照)
Input样本是片段化后未经抗体富集的染色质,通过去纯化处理得到的DNA。它充当实验的自身对照,用于提供基因组背景参考。
(1)在peak calling分析中,它作为背景参照,帮助识别IP样本中真正的富集峰。因为基因组某些区域可能自然存在reads堆积(如高GC区域),Input能排除这种非特异性噪音,确保富集信号源于抗体结合而非实验或生物学偏差。
(2)Input和IP样本在实验前期(如质量检测、建库和测序)是平行进行的独立样本,Input可用于确认片段化效果及富集效率,在数据分析阶段需整合两者测序数据,软件通过比较IP和Input的reads分布(如使用MACS2等工具),确定基因组上显著富集的peak位置,并输出最终结果用于后续功能分析。
3、IgG阴性对照
阴性对照理论上基本不会捕获到足量DNA,不建议进行建库测序。IgG样本是用来做阴性对照使用的,理论上该蛋白是捕获不到任何DNA序列的,那建库就不能成功,最终测序也没有任何意义。非特异性结合就说实验体系的问题了,我们磁珠会先处理的,做到IgG拉不下来DNA就是将IP体系做到很严苛的条件的。如果对非特异性的要求很高,可以送测IgG。
4、标签抗体对照
使用野生型样本或仅表达标签的样本作为阴性对照,排除标签本身引起的非特异结合,确保peak信号源于目标蛋白而非标签效应。
Q3:ChIP-seq的样本重复性设置。
生物学重复指的是对不同来源的独立样本(如不同细胞培养批次或动物个体)进行重复实验,而非技术重复(同一样本的多次处理)。从科学严谨性角度出发,建议设置2-3个生物学重复。
(1)提升结果可信度:生物学重复有助于消除单次实验的偶然性偏差,确保peak calling(峰检测)的可靠性。
(2)满足统计分析与发表要求:许多高分期刊明确规定实验数据需包含生物学重复。另外在分析的时候也会具有生物学的统计学意义,,这样不管是进行主成分分析还是热图聚类分析,都可以满足要求。
(3)从目前文章接收的角度来说,可以设置2个重复(支持多数期刊),若资源充足,3个重复结果更为可信(符合高分期刊接收标准)。但如果经费紧张,可考虑折中方案:先进行一次ChIP-seq实验,再通过ChIP-qPCR等验证性实验验证关键靶点(设置重复作为补充验证)。
Q4:想设置2次或者3次生物学重复,但是想先送一次样本看看情况,然后再送其它生物学重复是否可以?
不建议这样操作,可能会存在批次效应影响。不同批次的样本可能因储存温度、运输时长或试剂批次变化(如抗体效价波动)导致数据不可比。在ChIP-seq中,这会放大背景噪声,使后续重复的数据难以通过一致性检验,甚至筛选不到显著差异peak。生物学重复的核心前提是样本处理的同质性(即所有重复在相同条件下并行处理)。分批次操作破坏了这一原则,可能被审稿人质疑结果的可靠性,增加论文返修风险。为确保实验成功,所有生物学重复的样本应一次性采集、处理和运输,保持实验条件(如交联时间、抗体孵育)严格一致。如果在资源分配上需分步进行,可考虑使用“冻存样本”策略:预先制备并冻存足够样本,后续在相同实验周期内完成重复处理,以最小化批次变异。
Q5:ChIP-seq的样本的取样时期和部位怎么选择?
1、组织/部位选择:具体是根据您的研究需求确定的,若关注特定生理状态(如胚胎发育、肿瘤进展),需选取疾病相关或功能活跃的组织(如脑组织研究神经分化、癌组织研究驱动蛋白)。不同组织存在修饰的差异,可能会影响蛋白和DNA的结合。在实验开始前,建议通过Western Blot或免疫组化预实验确认目标蛋白在候选组织中的丰度(确认目的蛋白表达情况,避免因蛋白过低导致富集失败)。
2、时间点选择:优先选择靶蛋白发挥核心功能的时期。对动态过程(如细胞周期),建议设置多个时间窗口,避免单一时间点遗漏关键结合。
(1)发育研究:需覆盖关键形态发生阶段(如小鼠胚胎E11.5神经管闭合期)。
(2)药物处理:依据药代动力学确定峰值作用时间(如激酶抑制剂处理后2-4小时)。
3、特殊样本注意事项
(1)原代细胞:需控制传代次数维持表型。
(2)临床样本:冻存组织应速冻于液氮,避免染色质降解。
(3)多细胞类型混合:建议先分选特定细胞群(如用FACS分离T细胞),防止信号稀释。
Q6:ChIP-seq的样本分组设置。
ChIP-seq实验中,样本分组设置是关键步骤,直接影响结果的可靠性和生物学意义。分组应根据研究目标(如聚焦特定表型或功能)灵活设计。
1、单组样本设计的适用性与局限性
如果只使用单一细胞组(如仅肝癌细胞),该方案能鉴定样本在当前状态下的目的蛋白全基因组调控位点。然而,这种方法可能包含与研究表型相关的位点,同时也混杂无关位点。为提升筛选效率,建议结合RNA-seq等转录组数据,通过联合分析优先识别与基因表达变化相关的调控位点。
2、对照组设计以增强结果的稳健性
更可靠的设计是引入对照组(如肝癌细胞 vs. 正常肝细胞),通过比较识别差异结合峰。例如,在肝癌研究中,正常细胞作为基准,癌细胞中显著变化的峰(如增强或减少结合)可直接关联疾病表型,提高目标位点的特异性。
3、单一处理组仅针对特定蛋白全面扫描全基因组结合位点,适用于基础图谱构建。设置条件分组更适合聚焦功能表型(如发育或应激响应),建议设置对比组(如不同发育阶段、逆境处理前后、或表观遗传修饰差异样本),以此生成差异结合数据,在结题报告中重点展示差异分析部分(如热图或富集通路),以揭示表型相关的动态调控机制。
Q7:实验开始前WB质检的目的是什么?
在启动正式的ChIP-seq实验流程之前,进行Western Blot质)是至关重要且不可省略的预实验步骤。
(1)验证抗体特异性与结合能力:确认选用的抗体能否特异性地识别并有效结合目标蛋白(抗原)。通过WB结果,重点观察目的蛋白条带(预期大小)。理想情况下,目的条带应为最明亮的主带。需要评估是否存在非特异性结合产生的杂带,以及这些杂带的强度相对于目的条带的比例。如果杂带过多或强度过高,表明抗体特异性不佳,将严重影响后续ChIP实验的特异性和结果可靠性。
(2)评估目标蛋白的表达水平:检测在计划用于ChIP的实验样本(如特定细胞系或组织)中,目标蛋白是否存在表达及其相对丰度。WB信号强度(目的条带亮度)直接反映了样本中目标蛋白的基础表达量。这对于判断样本是否适合进行ChIP至关重要。如果蛋白表达量过低,可能低于ChIP实验体系(包括后续公司质检)的检测下限,导致免疫沉淀失败或信号微弱,无法获得有意义的测序结果。
Q8:瞬转细胞系是否可以做?
不推荐作为首选方案,尤其对于ChIP-seq这种需要高稳定性和一致性的实验,存在显著风险,包括蛋白表达不稳定、转染效率低和样品异质性等问题。
(1)瞬转的蛋白表达通常波动较大,而稳转细胞系通过筛选和克隆化,能实现长期稳定的高表达水平,能提供更强的信号强度和实验可重复性。相比之下,瞬转依赖瞬时质粒表达,如果错过表达峰值,实验结果极易偏差。
(2)瞬转的转染效率不能确定,且无法保证所有细胞均成功表达目标蛋白。Western Blot等只能评估总蛋白水平,无法区分单个细胞的转染状态,样品中混杂未转染细胞会降低目标蛋白的富集效率。
(3)ChIP实验样本需求量大,转染试剂毒性可能导致细胞死亡,批次间一致性差。
Q9:ChIP-seq测序量多少合适?
一般测序深度≥ 3×基因组覆盖深度可满足大多数常规ChIP-seq实验基因组覆盖。我司目前包含的标准测序数据量为 6 Gb,此数据量在绝大多数情况下足以获得高质量的峰值(peak)检出和可信的富集分析。 针对特殊情况:
(1)超大基因组物种:对于基因组显著大于常见模式生物的物种,3X深度对应的实际数据量将远超6Gb。此时,6Gb数据量可能不足。
(2)复杂或弱富集目标: 若研究目标为弱富集蛋白或涉及复杂调控区域等,可能需要更高的测序深度和相应的更大数据量。
——建议在项目启动前与我们详细沟通您的具体研究物种基因组大小及目标蛋白特性等,进一步确认需求数据量。
Q10:ChIP-seq的生信分析流程是什么?
(1)原始数据质控与预处理:使用FastQC评估测序数据质量(碱基质量分布、GC含量、N碱基含量)。去除低质量碱基和接头序列等,生成clean reads。验证数据量是否达标,确保测序深度满足后续分析需求。
(2)参考基因组比对:将clean reads比对至参考基因组。质控比对率、非冗余比对率等指标。
(3)富集区域识别(Peak Calling):蛋白质-DNA结合位点会富集大量DNA片段,形成显著高于背景噪声的 “峰(Peak)”。使用对照样本排除背景噪音。
-
转录因子:结合位点明确,呈现高而窄的尖峰。
-
组蛋白修饰:结合区域广泛,表现为宽峰(Broad peak)。
(4)Peak注释与功能关联:
通过基因组注释将Peak关联至最近基因的启动子区(TSS)、内含子/外显子、增强子等功能区域。
并靶基因功能富集对Peak相关基因进行GO、KEGG等富集分析,揭示相关生物学功能。
(5)Motif分析与差异分析:在Peak序列中鉴定规律性转录因子结合基序。多样品比较时,进一步分析识别组间差异结合位点。后续可通过IGV 或 UCSC Genome Browser 直观展示具体关注感兴趣基因Peak分布与测序深度。

图 ChIP-seq生信分析流程。
部分分析结果展示



















 4万+
4万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









