







一、文件准备
1.下载fasta文件或者你所研究的物种的蛋白质序列文件
从NCBI下载的或者通过提取DNA/RNA或送测序得到的数据
2.查阅文献或通过参考同源基因,去Pfam数据库下载相应的hmm结构文件,这里下载的是PF00248.hmm
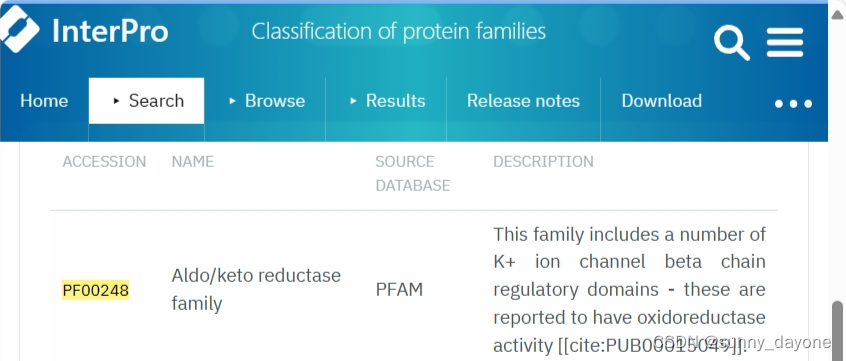
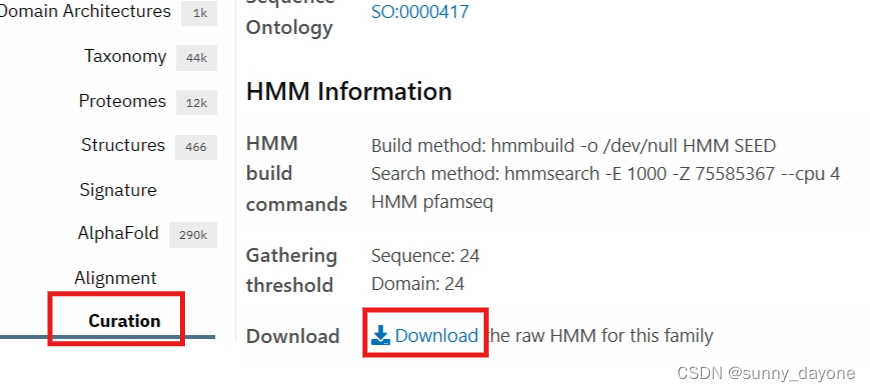 3.下载hmmer软件
3.下载hmmer软件
网盘:(后续所需的TBtools、MEGA也在网盘里)
链接:https://pan.baidu.com/s/1oqcn-lN19tkXUn5kjKdcFQ?pwd=xixh
提取码:xixh
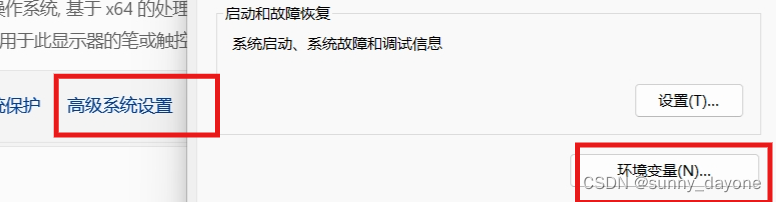
4.解压后把文件的安装路径加入到环境变量中
win11右键“此电脑”——“属性”——“高级系统设置”——“环境变量"
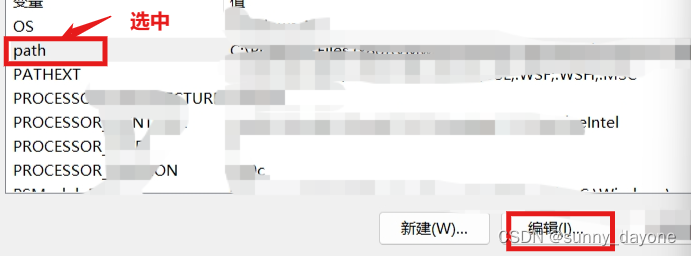
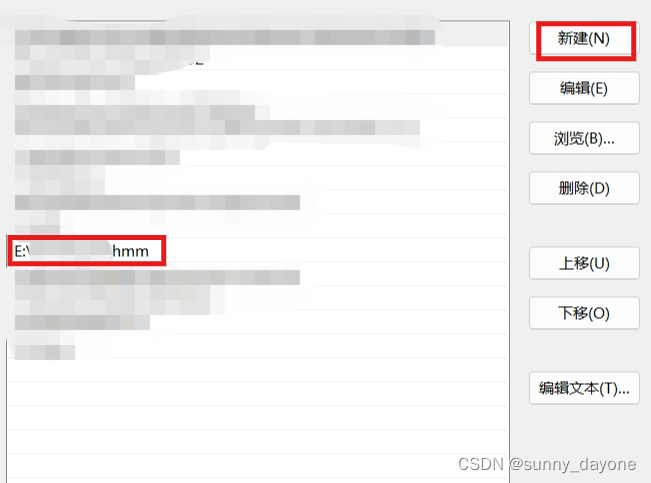
点击”确定“
5. hmm检索

打开cmd:“shift+鼠标右键”——”终端管理员“
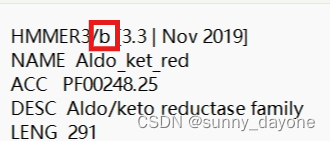
cd 所有文件所在路径/ #建议把以上所有文件放在同一个文件夹里面,方便操作用记事本打开下载的hmm文件(PF00248.hmm),将f改为b
hmmsearch.exe -E 1e-5 .\PF00248.hmm .\protein.faa > out.txt #1e-5为筛选值将out.txt 中的ID号提取出来,保存为Excel文件或者txt文件
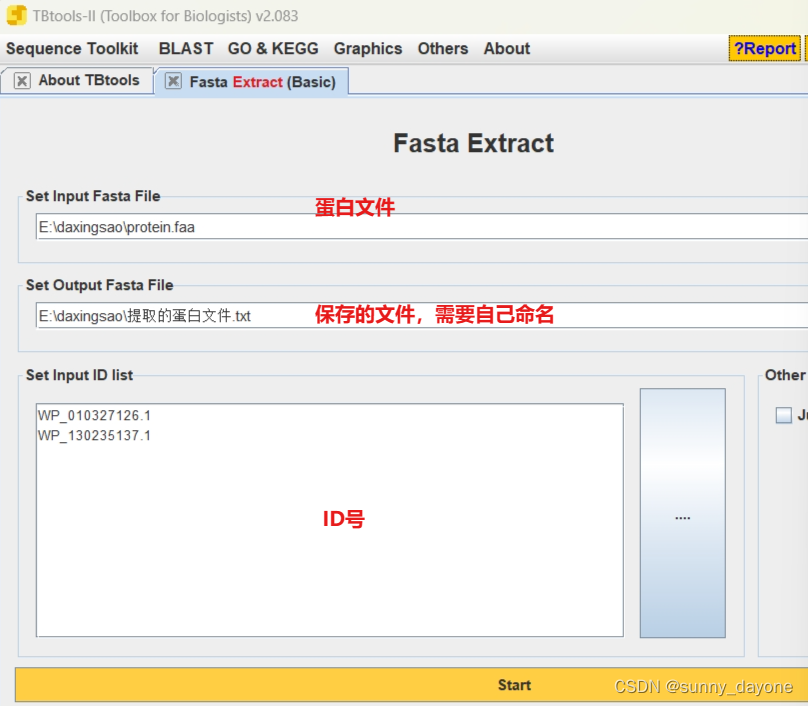
6.利用TBtools通过ID号把这些蛋白序列从物种的蛋白质序列中提取出来
Sequence Toolkit —— Fasta Tools —— Fasta Extract(Basic)
#注意复制ID号后把最后的空行删除
7.文件合并
用记事本打开保存的文件,全选复制
Ctrl+A Ctrl+C
用记事本打开下载的超家族文件(.fasta文件)
Ctrl+V
二、多序列对比
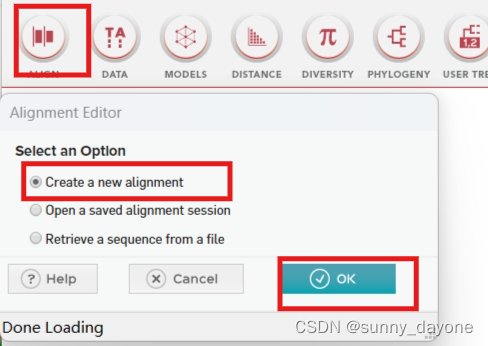
”ALIGN"——“Edit/Build Alignment" ——"Create a new alignment"——"Protein"
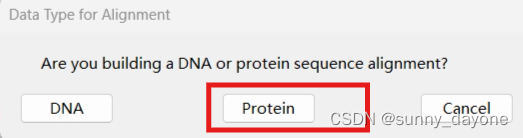
点击”Edit“——"Insert Sequence From File"导入我们需要比对的序列(需拉到最后,检查有无空行,若有,鼠标右键——delete)
ctrl+A全选,点击 Muscle(肌肉图标,“W”指的是ClustalW算法,若发文章建议选择ClustalW算法)
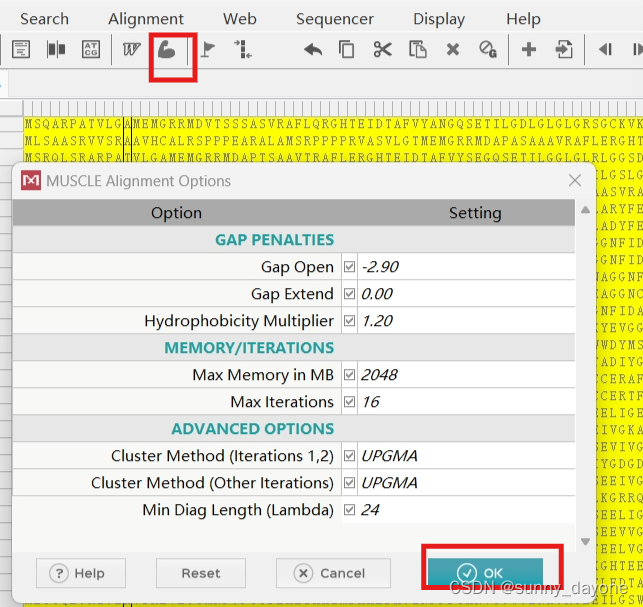
"Data"—— "Save Session"保存序列比对的结果
三、构建系统发育树
"Data"——"Phylogenetic Analysis"进行系统发育分析
使用最大似然法建树前先进行测试,再选用模型
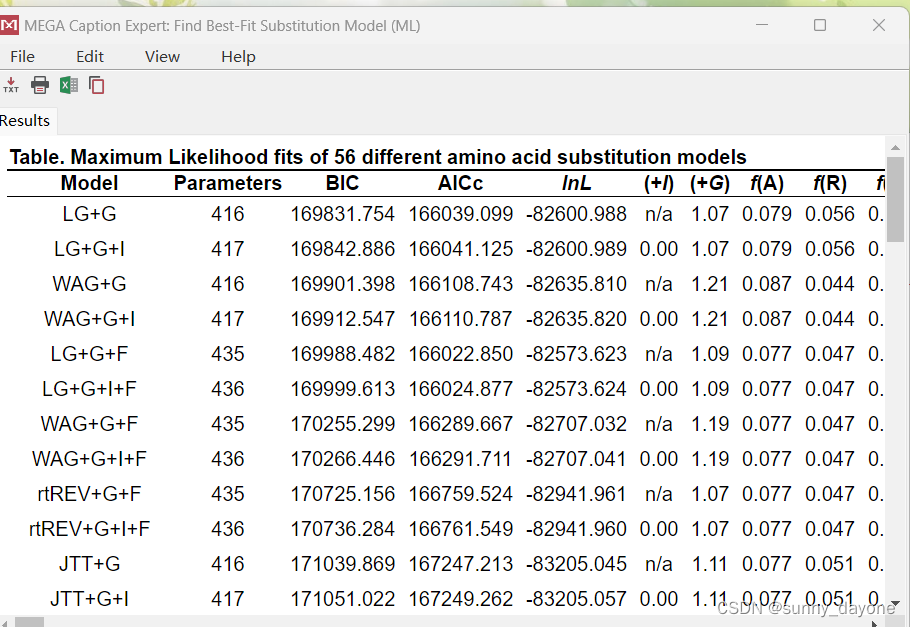
“MODELS”——“Find Best DNA/Protein Models(ML)..."(漫长的等待...)
结果如下:
”PHYLOGENY“——"Contrust/Test Maximun Likelihood Tree..."
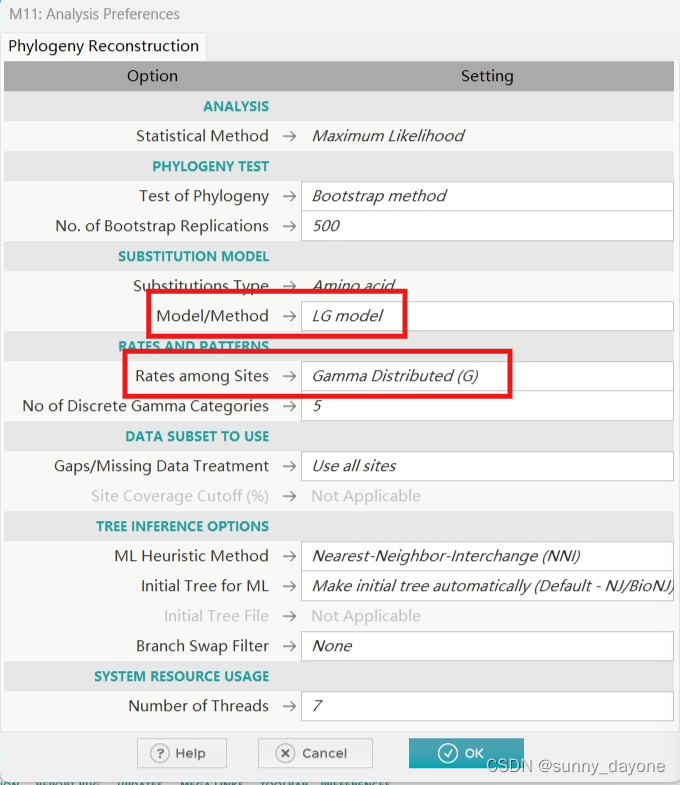
(更加漫长的等待...)

















 998
998

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









