







一、准备物种的DNA fasta/faa/fas/fa文件和gff/gff3文件
二、调整序列ID(避免不同物种序列 ID 冲突问题)
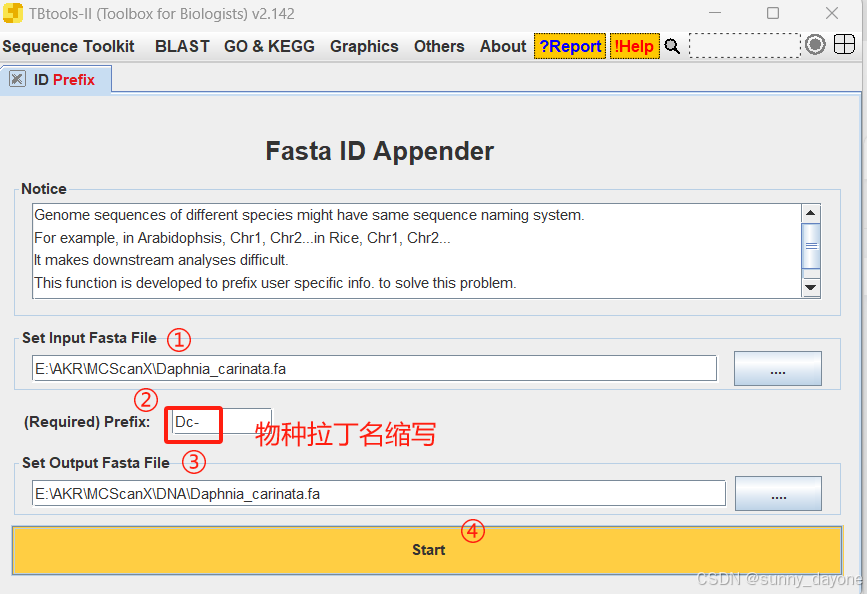
1.加前缀
Sequence Toolkit ---Fasta Tools ----ID Prefix (fasta文件)
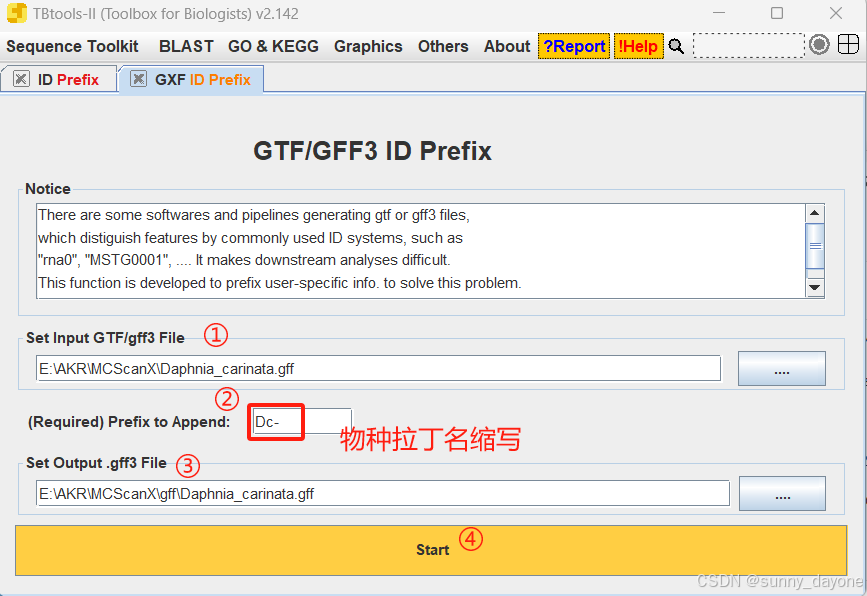
Sequence Toolkit ---GFF3/GTF Manipulate ---GXF ID Prefix
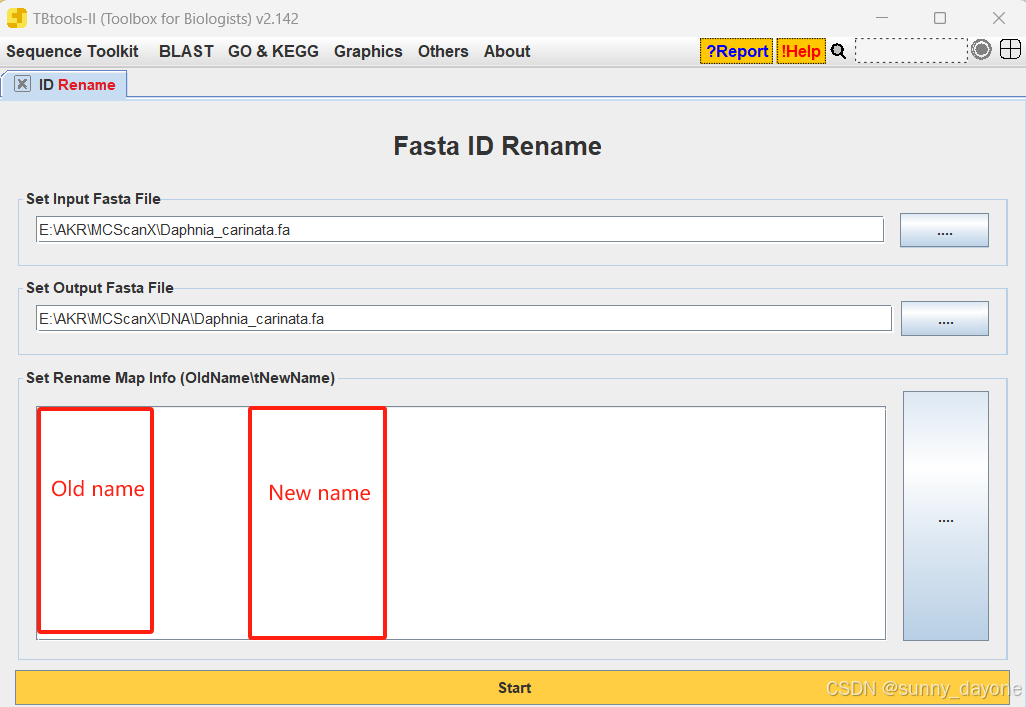
2.或者对序列ID进行重命名
Sequence Toolkit ---Fasta Tools ----ID Rename
Sequence Toolkit ---GFF3/GTF Manipulate ---GXF Rename
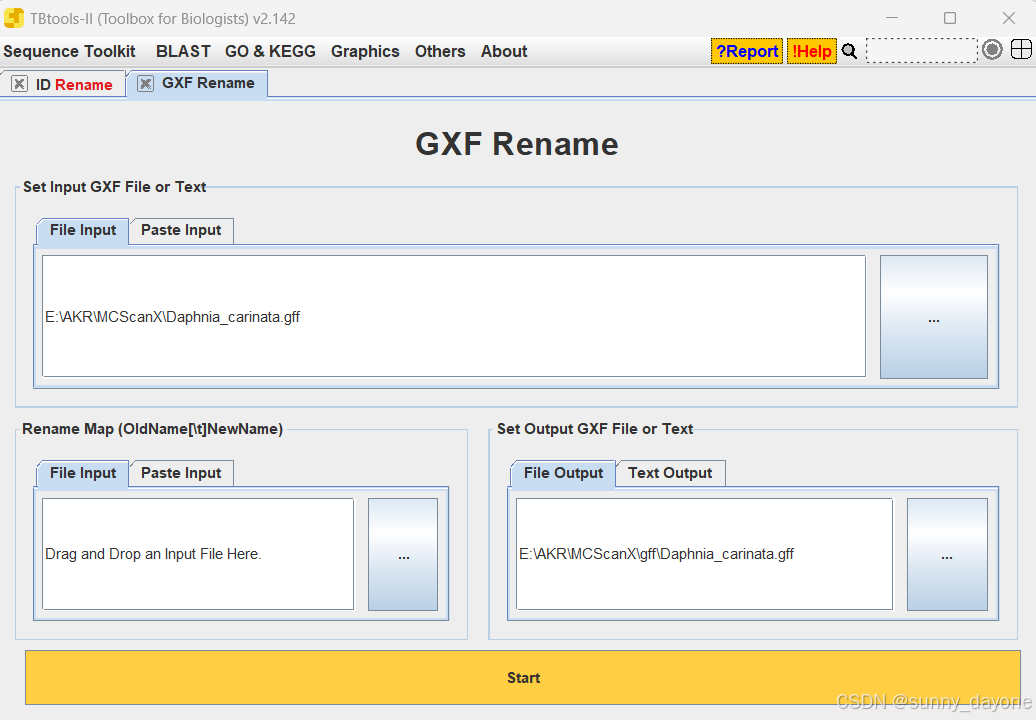
##Rename 文件格式:旧ID[制表符]新ID
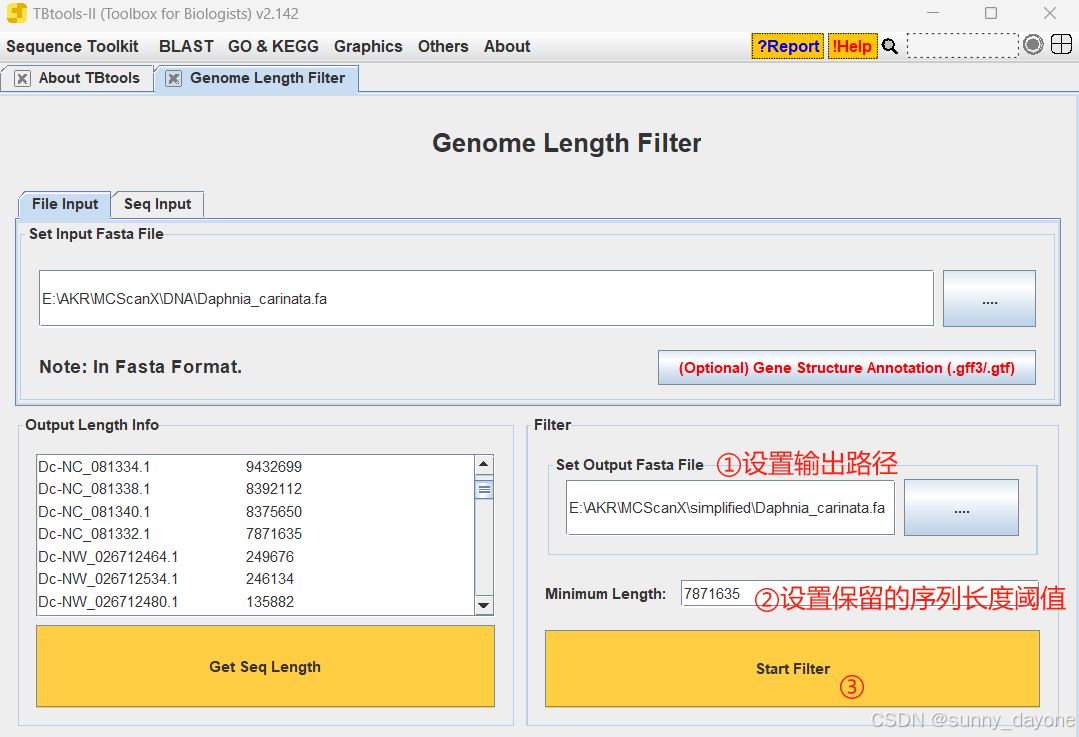
三、过滤基因组序列和注释碎片
进行共线性分析时,图上总是有很多黑块,很乱,如下图,是因为参考基因组中染色体碎片太多,我们需要去除这些碎片
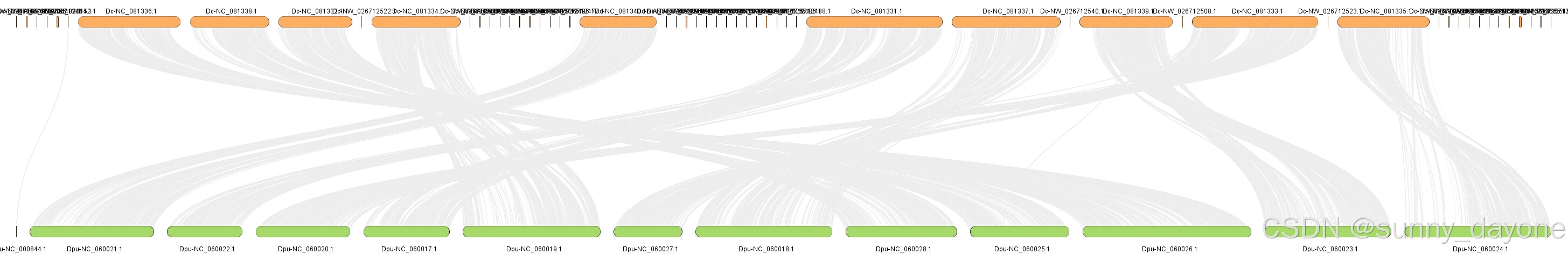
Graphics---Comparative Genomics---Genome Length Filter
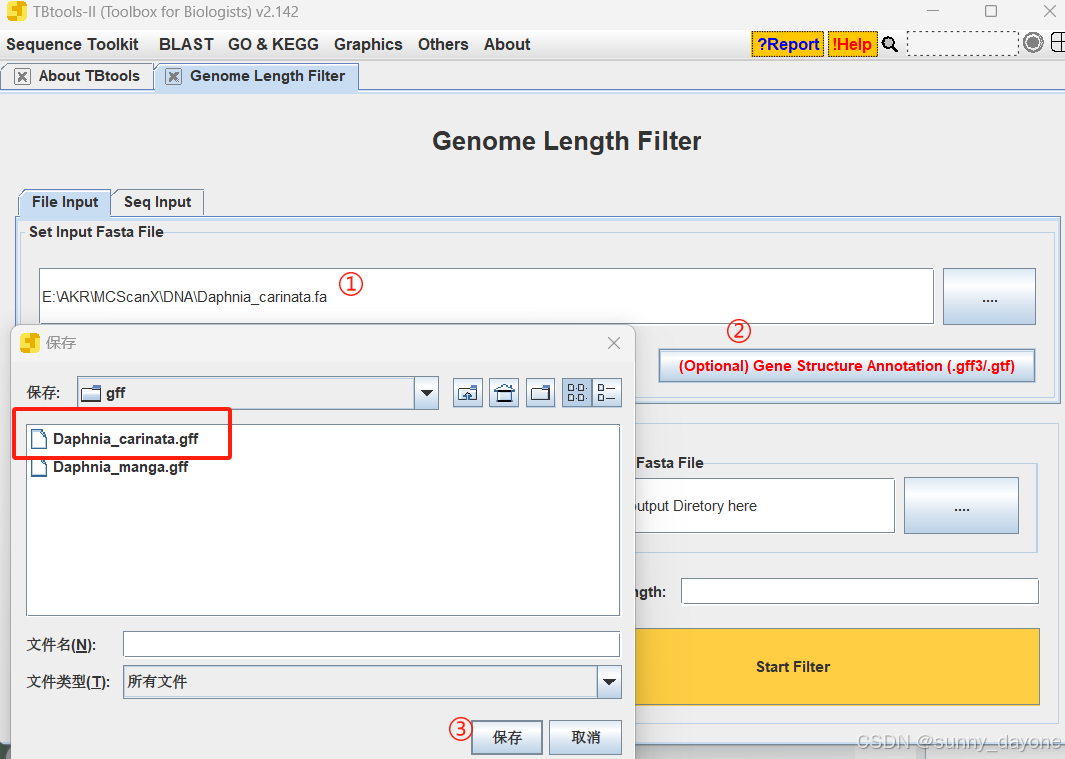
点击"Get Seq Length"获取序列长度
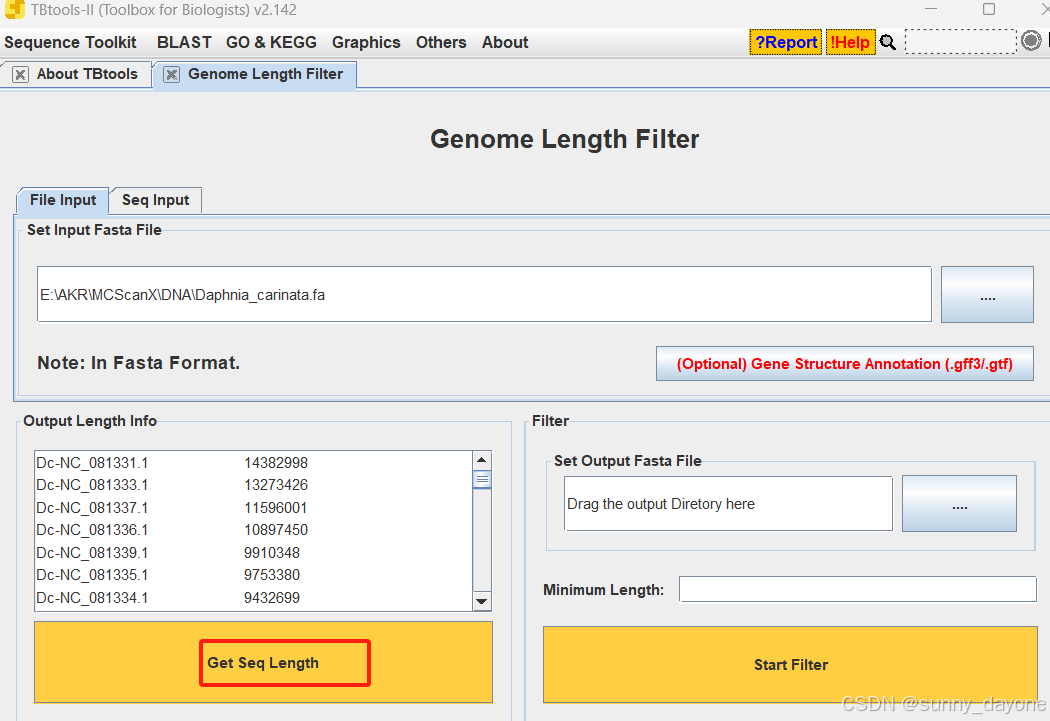
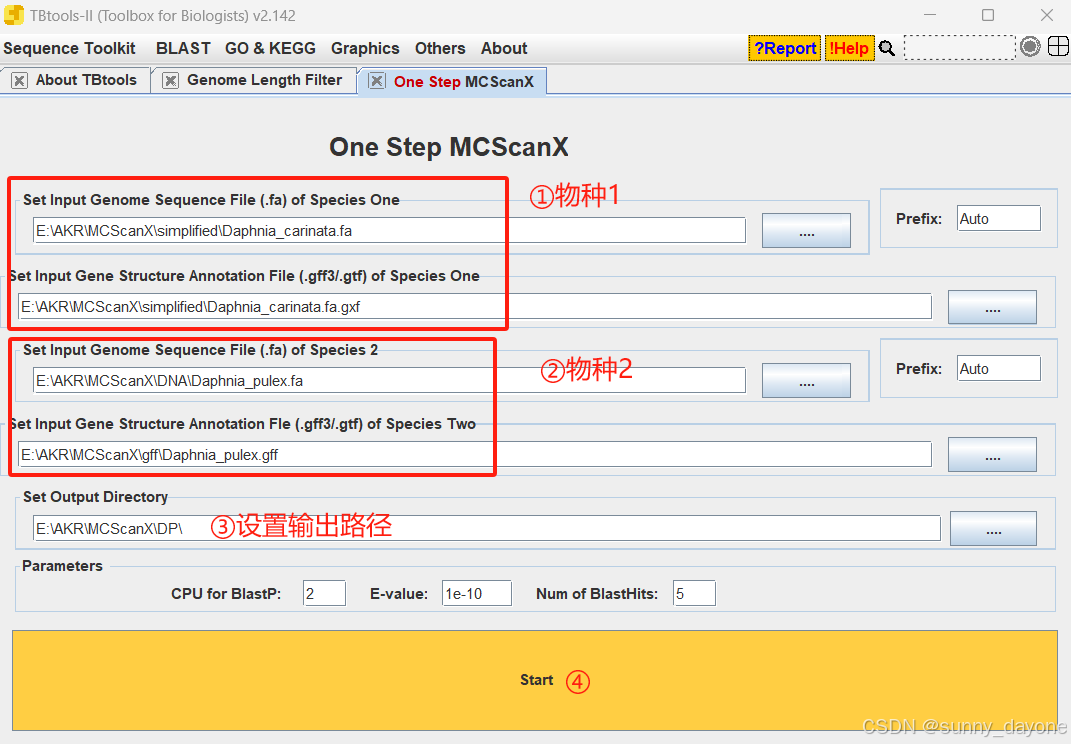
四、物种两两间进行共线性分析
Graphics---Comparative Genomics---One Step MCScanX
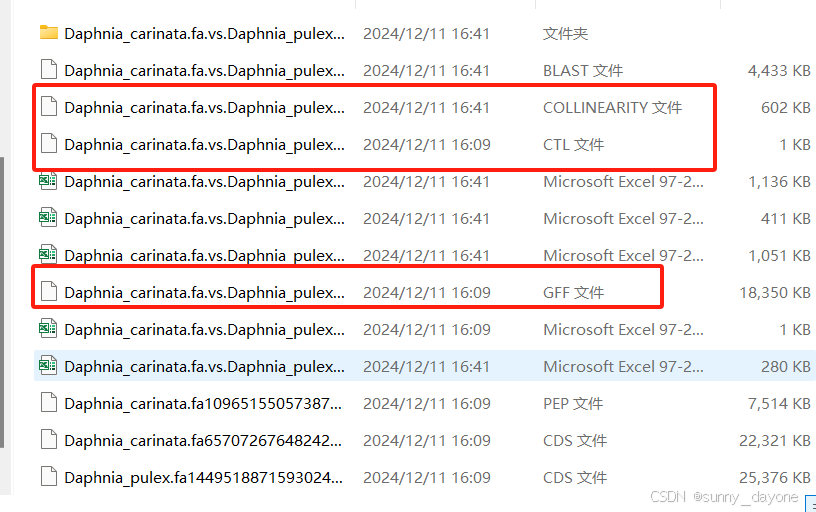
结果
可视化
需要结果文件中的COLLINEARITY文件、GFF文件和CTL文件
Graphics---Comparative Genomics---Dual Systeny Plot ffor MCscanX
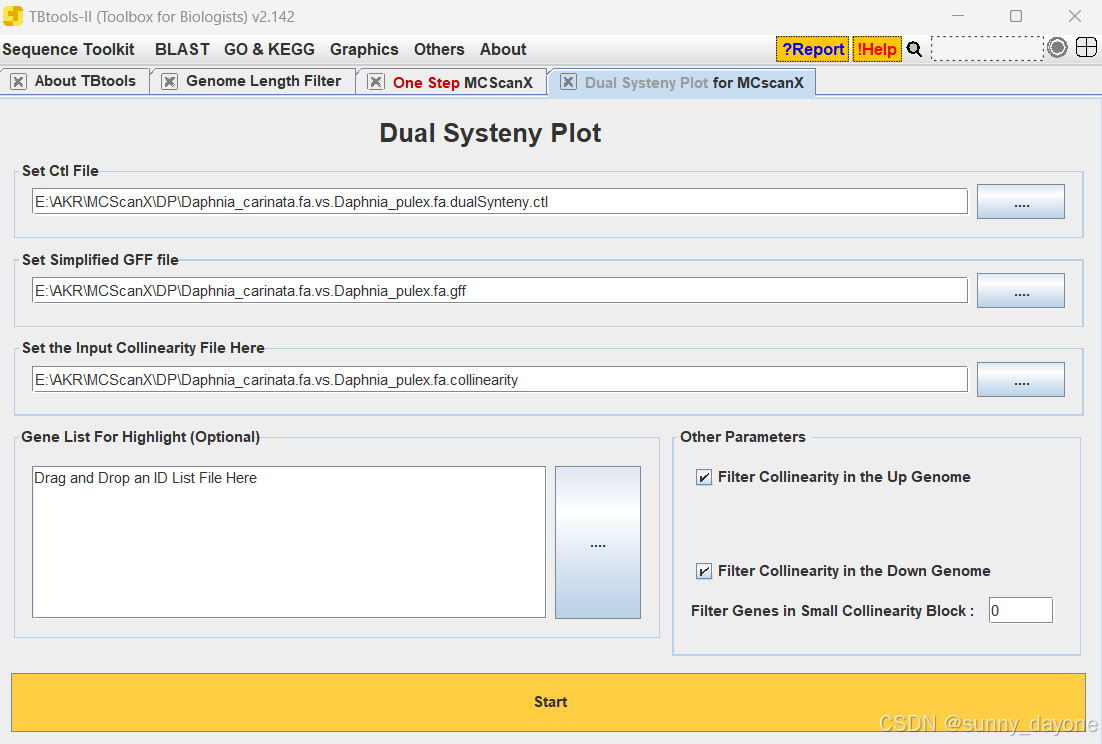
“Gene List For Highlight”(可选)填希望高亮显示的基因ID,这里没有填写,结果如图:
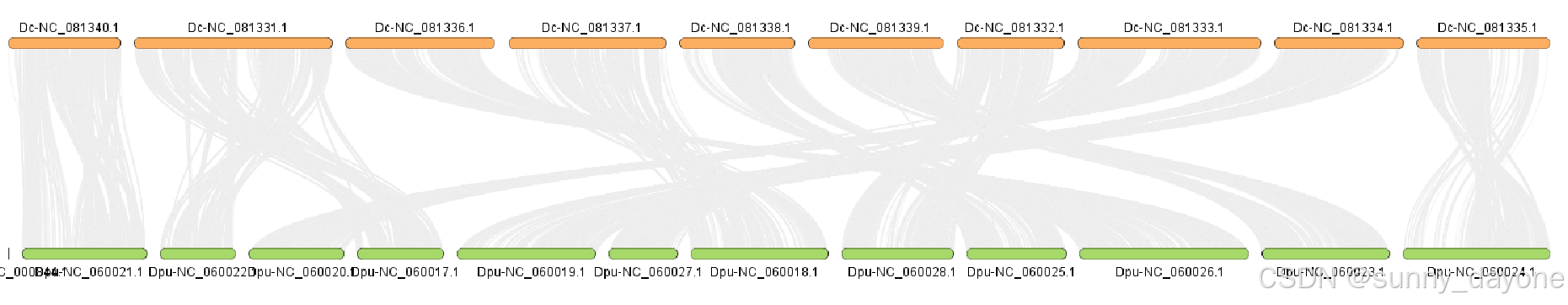
若只做两物种间的共线性分析,到此就可以了,多物种间的共线性分析可以继续往下看
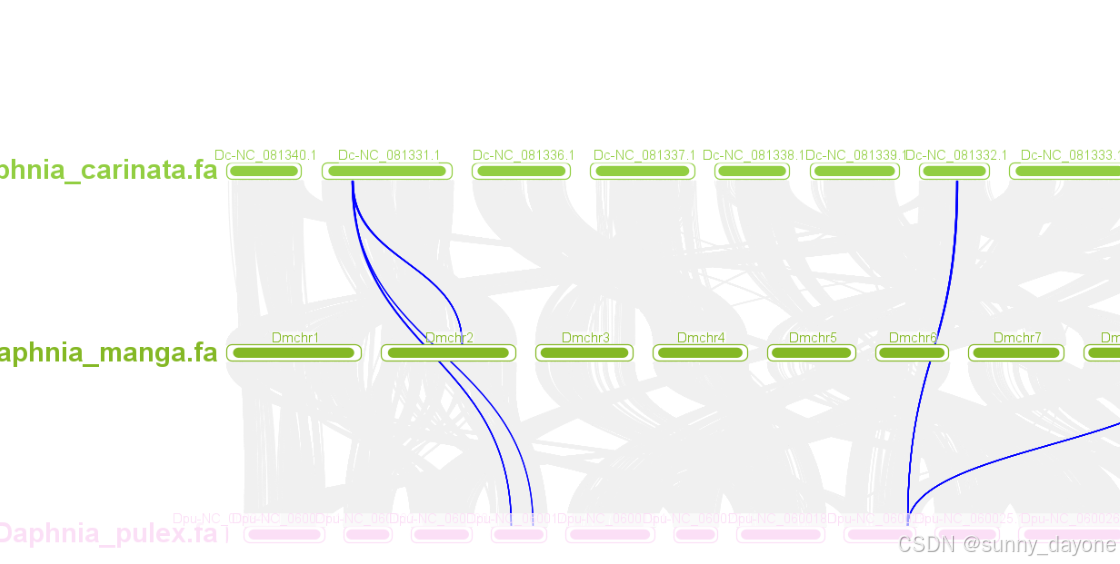
五、多物种间共线性分析
Graphics---Comparative Genomics---Unlimited Synteny Visualization即可,但我运行时报错,故另择他法
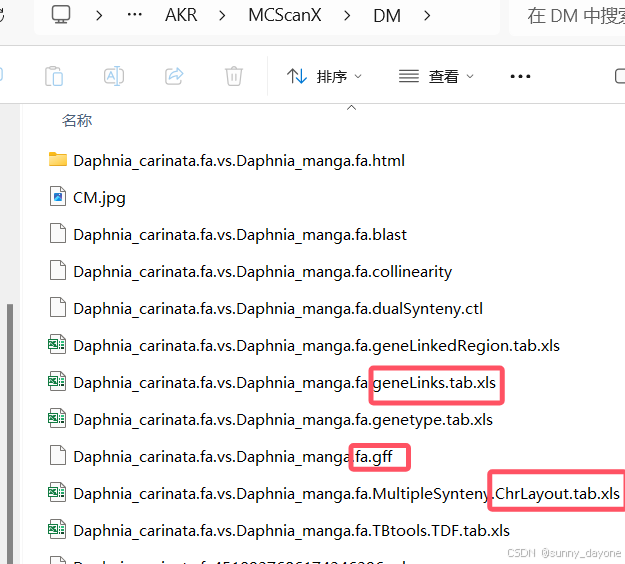
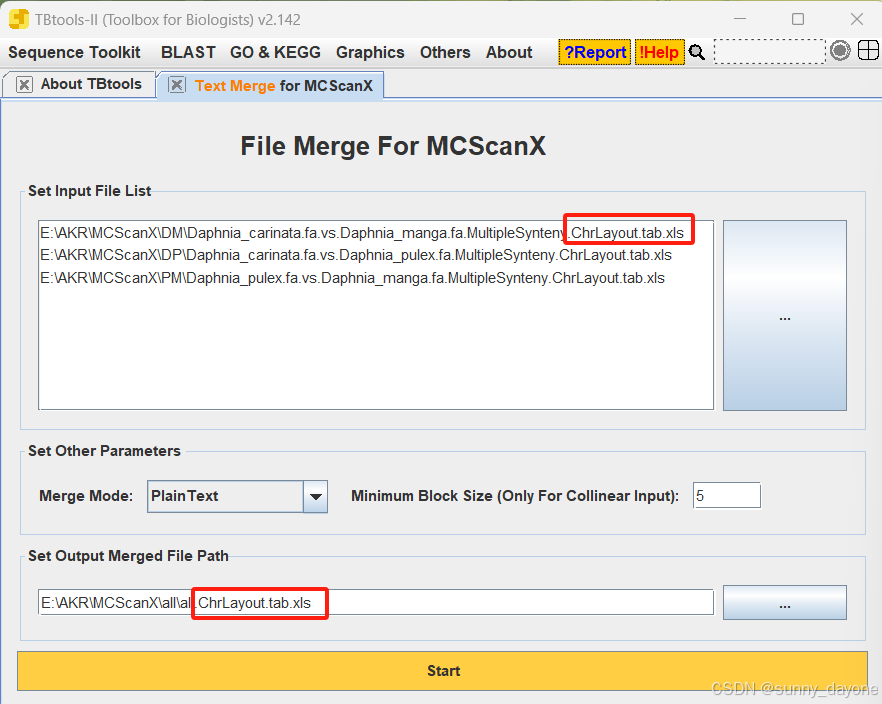
1.合并
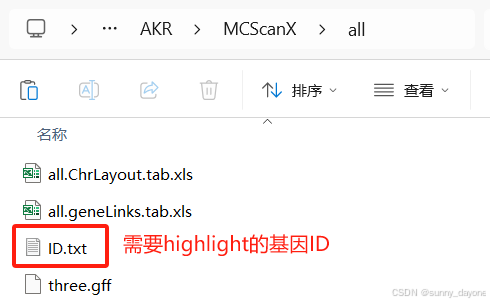
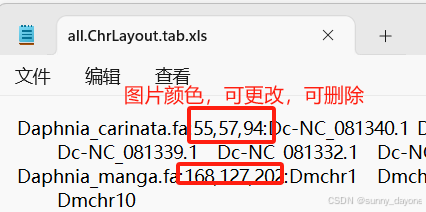
需要第四步两两共线性分析结果文件中的gff文件、.ChrLayout.tab.xls文件和.geneLinks.tab.xls文件(如图)
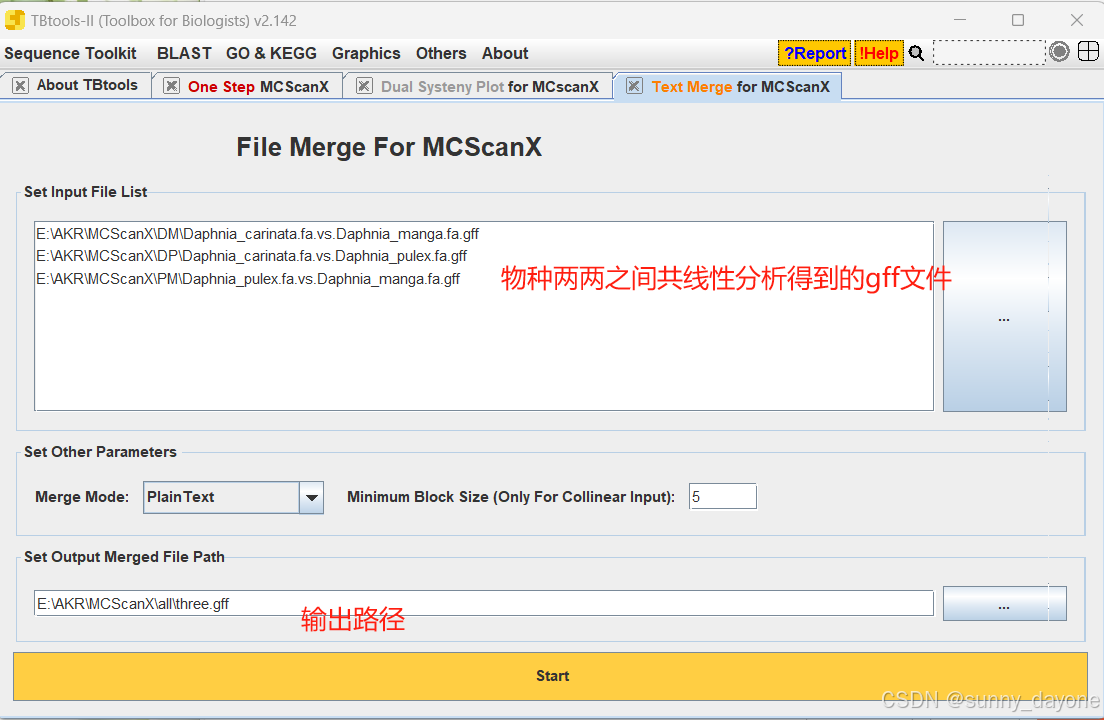
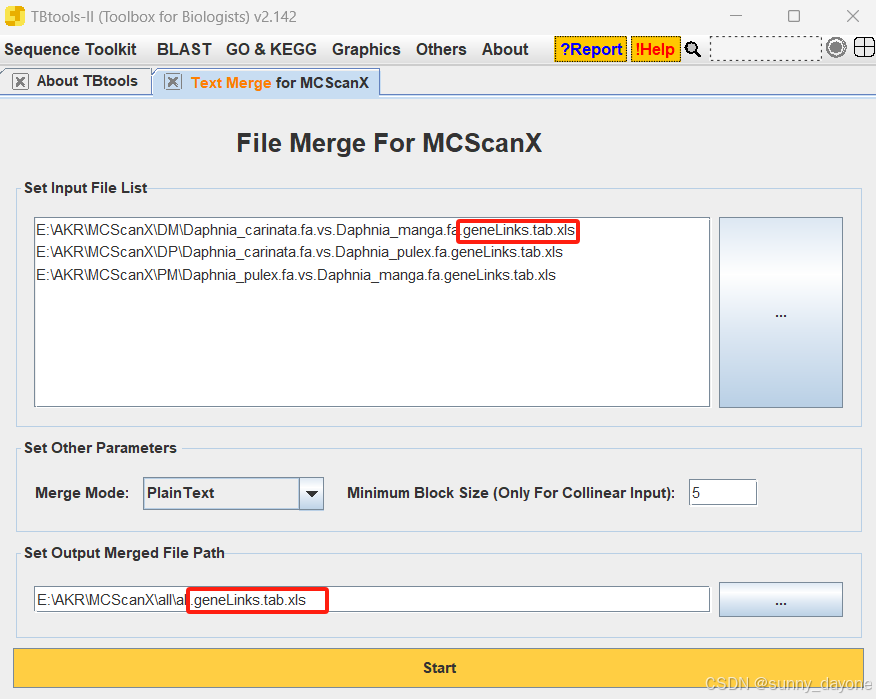
Graphics---Comparative Genomics---Text Merge for MCScanX
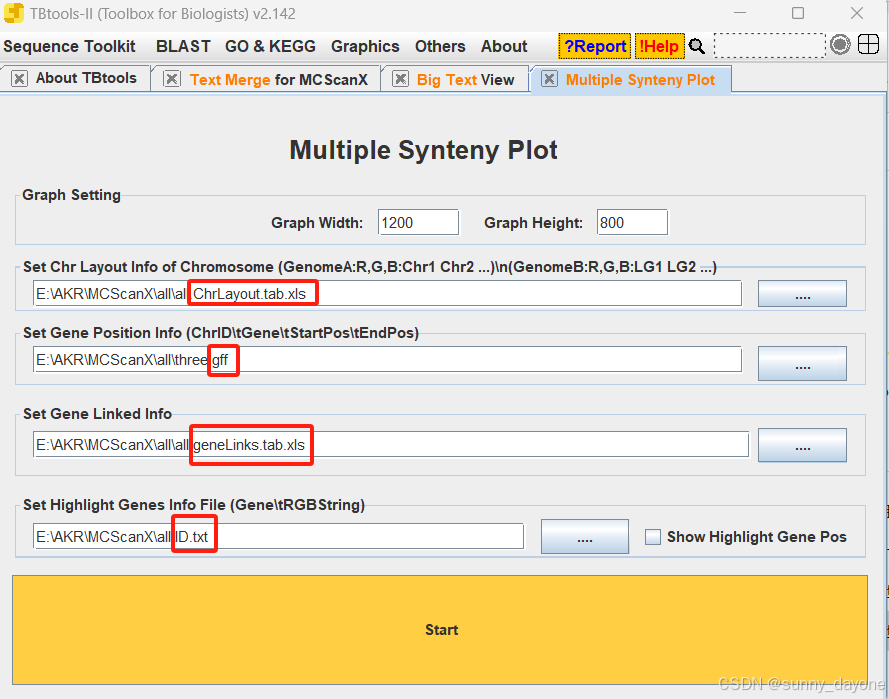
2.可视化
Graphics---Comparative Genomics---Multiple Synteny Plot

















 3万+
3万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









