




 神经退行性疾病如阿尔兹海默症、帕金森症等目前无有效治愈方法。然而,干细胞疗法展现出潜力,有望修复受损神经元。本文以亨廷顿病为例,探讨干细胞疗法的进展,包括神经干细胞、胚胎干细胞和诱导性多功能干细胞的使用,以及面临的挑战和未来前景。
神经退行性疾病如阿尔兹海默症、帕金森症等目前无有效治愈方法。然而,干细胞疗法展现出潜力,有望修复受损神经元。本文以亨廷顿病为例,探讨干细胞疗法的进展,包括神经干细胞、胚胎干细胞和诱导性多功能干细胞的使用,以及面临的挑战和未来前景。
关注“心仪脑”查看更多脑科学知识的分享。
关键词:脑科学、干细胞疗法、行业动态
神经退行过程(neurodegeneration)指神经元结构或者功能的丧失,可能最终会引起神经元死亡。
常见的神经退行性疾病包括阿尔兹海默症、帕金森症、亨廷顿病(Hungtinton’s Disease, HD)、多发性硬化(MS)、渐冻症(ALS)。神经退行过程可以在不同层级的神经元回路中发生。现有药物可以暂缓表面症状(如亨廷顿病的非自愿性动作),但没有药物和疗法被证实可以在人体中预防或者逆反神经退行的过程,所以神经退行性疾病通常被认为是无法痊愈的。
但近年来,干细胞疗法被发现有望修复受损的神经元组织并替代受损神经元,以改善神经退行性疾病的病情。本文将以受关注还较少的亨廷顿病 (HD) 为例,聊聊干细胞疗法所能带来的潜在希望以及瓶颈。
01介绍一下亨廷顿病与干细胞
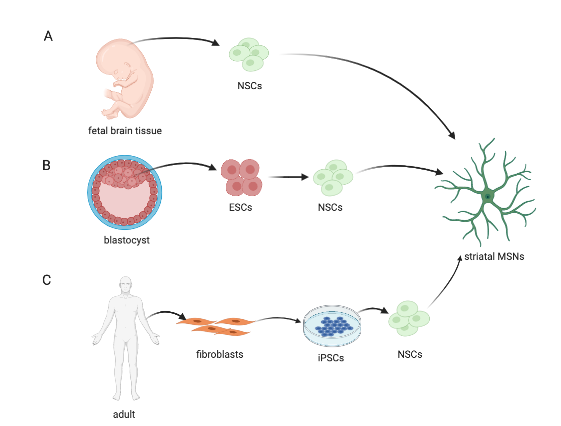
亨廷顿病HD是由于HTT(huntingtin)基因变异引起的。HTT基因中的CAG序列长度通常为35,而变异的HTT基因中CAG的长度可达100。这一变异使得神经元细胞,特别是纹状体中的GABA能中等棘神经元(medium spiny neuron, MSN)变得脆弱,致使基底节通路(basal ganglia pathway)功能障碍,引起行为及认知异常。
干细胞(stem cell)是一种未分化或部分分化的细胞,处在细胞谱系的最早阶段。它可以在生物体内或体外分化为至少一种类型的高度分化的细胞。
HD的治疗涉及到过三种干细胞:神经干细胞(neural stem cell,NSC)、胚胎干细胞(embryonic stem cell, ESC)、诱导性多功能干细胞(induced pluripotent stem cell, iPSC)。
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1091
1091

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









