



日常科研中,ChIP-qPCR是解析蛋白与DNA互作的“利器”——它既能高效验证ChIP-seq的筛选结果,排除假阳性数据,也能直接检测已知蛋白(如转录因子、组蛋白修饰)与特定DNA序列的结合情况,为基因调控机制研究提供关键证据。而引物设计作为ChIP-qPCR实验的关键前提,直接影响扩增效率、特异性与最终数据可信度,是每个科研人必须攻克的核心环节。今天小编就结合实验经验,带大家系统梳理ChIP-qPCR引物设计的方法与技巧。
从理论层面看,ChIP-qPCR的引物设计与常规qPCR设计原则一样。比如引物长度17-25bp(避免非特异结合或扩增效率下降),GC含量:40-60%(45-55%理想),正反引物Tm值差异不宜过大(保障扩增同步性),碱基分布尽量均匀等等。但核心差异在于,ChIP-qPCR的检测对象是“经ChIP实验富集的DNA片段”,而非基因组总DNA。这意味着引物设计的核心在于精准选择靶区域——需确保引物扩增的片段恰好覆盖目标蛋白的结合位点(如启动子区的转录因子结合元件、组蛋白修饰位点),才能准确反映蛋白与DNA的互作情况。接下来,我们将分几种不同方式详细拆解ChIP-qPCR的引物设计策略,帮你避开常见误区、提升实验成功率。
01 直接采用文献已发表的引物序列
根据前期已发表结果查找引物序列,是最简单且可靠的方案。这类引物经过实验验证,能有效降低实验误差。在文献中材料方法部分直接复用验证有效的引物序列。
02 基于前期测序结果
若文献中无现成引物,但有测序结果,可借助测序数据进一步筛选。
1、访问UCSC Genome Browser:http://genome.ucsc.edu/
2、选择测序生信分析所用的对应物种的参考基因组版本:在顶部导航栏点击Genomes,选择物种和版本(如人类选 GRCh38/hg38 或 GRCh37/hg19)(版本一定不能选错,否则序列不匹配)。
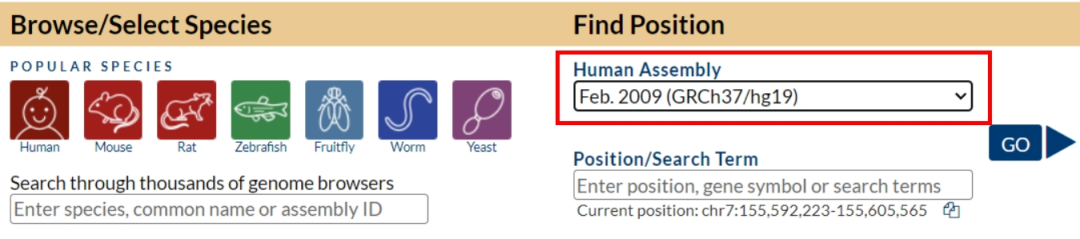
3、选择要查找的序列位置:在测序结果中选定具体peak,针对选定的peak可以直接复制其起点和终点,但是需要注意qPCR的目的扩增片段长度建议在200bp以内,选择的片段范围不能太长或太短,可以考虑以abs_summit(peak峰值)为中心进行前后延伸,上下游延伸长度可以不对称,尽量包含进peak里面,根据峰型进行调整。
4、将确定的peak的位置输入Position区域,如chr7:155,592,223-155,592,423(染色体编号:起点-终点),点击GO。
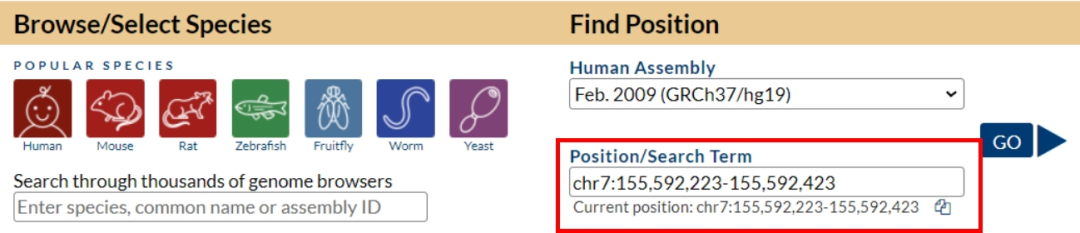
5、提取序列:点击顶部菜单View → DNA。
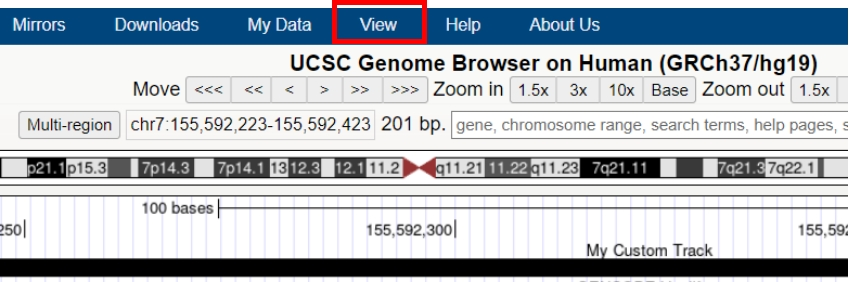
6、如为反义链需勾选Reverse Complement项(表示输出反义链互补序列,即原始反义链序列),点击Get DNA。
7、设计引物:生成序列后利用引物设计工具进行引物设计,目标长度150-200bp。
8、引物验证:得到引物后可以先用Input进行验证,确定引物特异性及扩增效率。
03 利用相关数据库测序结果
利用数据库中已发表的测序数据进行筛选,如人和鼠常用的数据库为Cistrome Data Browser,其收录了人与小鼠的已发表ChIP-seq数据,支持按物种类型、样本类型及因子进行精准筛选。
1、访问Cistrome Data Browser官网:http://cistrome.org/db/#/
2、填写转录因子名称、选择物种进行查询。
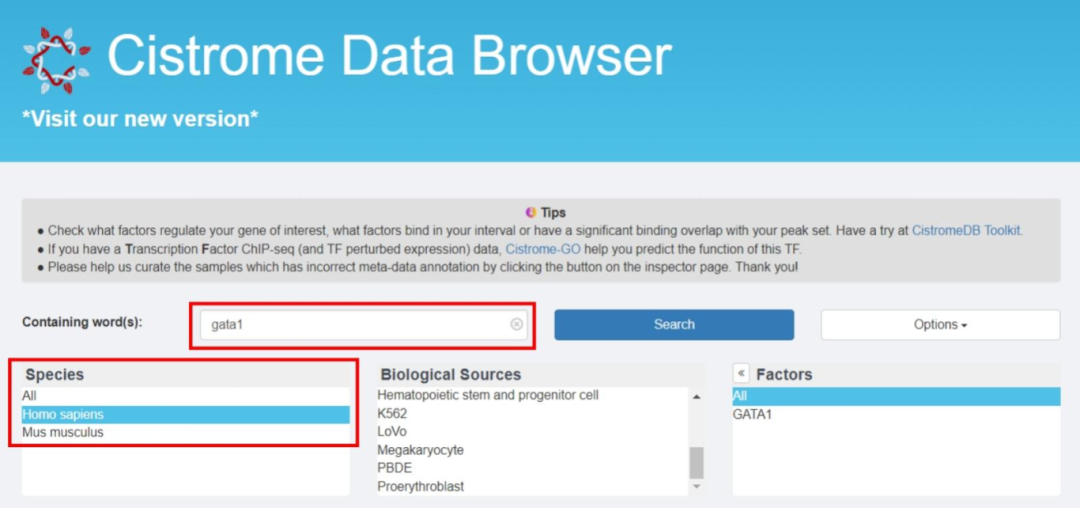
3、选择点击其中一种查询结果,可以看到项目的详细信息,还可以看到测序大致结果展示,点击UCSC Browser进入测序数据可视化界面。
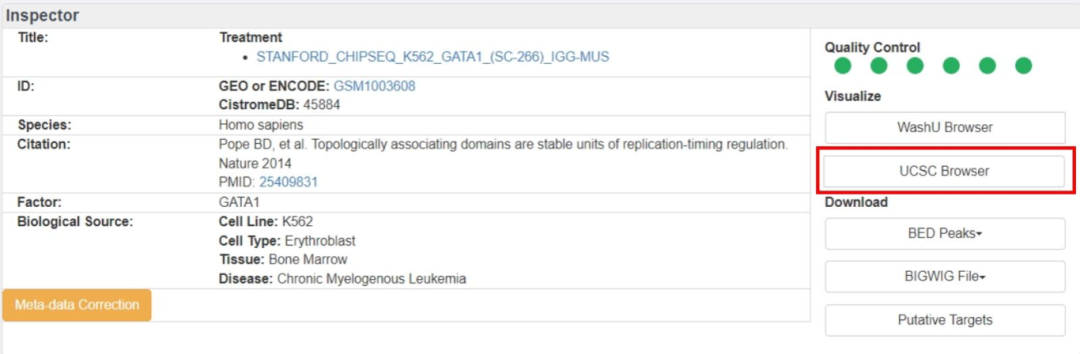
4、输入选择的特定peak,可以是具体位置或者基因名称,后续步骤同前。
04 利用保守性生信预测
对于常规模型物种,还可通过Jaspar等数据库预测转录因子结合位点,并在靶基因序列中筛选潜在互作区域。
1、明确核心信息:确定要研究的转录因子(比如“TP53”)和目标基因(比如“CDKN1A”)(物种:human)。
2、获取目标序列:转录因子通常结合在基因的启动子区域,我们需要获取目标基因转录起始位点(TSS)上游2000bp(这个范围是科研中常用的启动子核心区域,可根据研究需求调整)。序列可以从NCBI、Ensembl等数据库获取,注意保存为FASTA格式。
——小提示:获取序列时要确认物种版本(比如人类hg38、小鼠mm10),避免因版本错误导致结果偏差。
3、访问JASPAR数据库:https://jaspar.elixir.no/(数据库覆盖物种:脊椎动物、植物、昆虫、线虫等六大类)。
4、搜索目标转录因子:在搜索栏输入转录因子名称,选择物种,如果有其他需求可以在Advanced Options增加筛选条件。
5、从结果列表中选择目标motif,同一个转录因子小数点后面的数字越大表示版本越新。

6、勾选想要做分析的版本,点击右侧的Scan,将前面准备好的CDKN1A序列输入进去。匹配阈值默认80%(相似性百分比),严格预测可调至85%。点击 "Scan" 按钮运行预测。
(1)注意输入序列时格式标准FASTA格式(示例:>GeneX_promoter后换行输入序列)。
(2)长度建议:启动子区或其他<3Kb的序列范围。
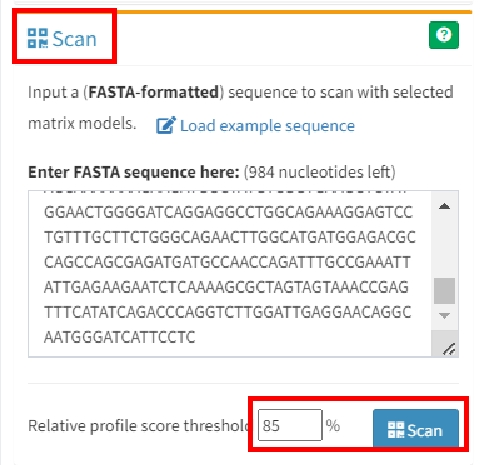
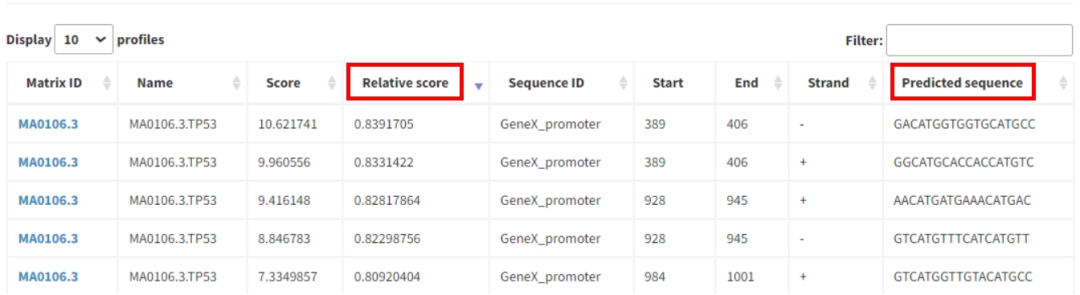
7、结果解读:
(1)相对评估值(Relative Score):0-100%,分值越高表明与已知motif匹配度越强(一般>85%为高置信)。
(2)预测位点(Predicted Sequence):结合位点碱基序列。
(3)注意如果为负链结合,在查找序列时为反向互补配对。
05 分段检测
如果无法确定具体的待验证结合区域,可以考虑对待验证区域进行分段设计引物,比如对一个较长的启动子区域进行验证,可以将整个区域划分为多个小片段,分别设计引物进行qPCR检测。
1、定位目标区域:获取基因启动子序列(或其他待验证序列)。
2、结合位点划分:将目标启动子划分为多个连续片段(如每段100-200bp),相邻片段重叠20-50bp,避免漏检。注意功能区域优先,包括转录起始位点、增强子、保守元件等。
示例:
| 片段编号 | 基因组位置 | 产物长度 | 设计要点 |
| 片段1 | -1000至-800 | 120bp | 覆盖预测结合位点 |
|
片段2 |
-850至-650 |
130bp |
与片段1重叠50bp |
|
片段3 |
-750至-500 |
120bp |
包含保守元件 |
3、使用引物设计工具设计引物,设计原则与普通qPCR原则一致。
4、引物特异性验证:
(1)用NCBI BLAST比对引物序列,排除非特异结合(如假基因或同源序列)。
(2)检查发夹结构和二聚体风险。
(3)使用基因组DNA做预实验,确认无扩增(排除DNA污染)。或者熔解曲线单峰提示特异性扩增。
小医叨叨
看到这里,相信你已经掌握了ChIP-qPCR引物设计的核心逻辑与实操技巧——但即便引物设计精准,ChIP-qPCR实验的成功还依赖样本交联的时效性、抗体的特异性、免疫沉淀的效率等多个关键环节,任何一步的疏漏都可能导致实验结果不理想,浪费宝贵的时间与样本。作为深耕蛋白与DNA互作研究服务的专业团队,我们的ChIP-qPCR服务为您提供全流程解决方案。无论您是需要验证ChIP-seq筛选的候选位点、检测特定转录因子与靶基因的结合情况,还是面对复杂样本(如稀有细胞、组织样本)不知如何入手,我们都能根据您的研究需求量身定制实验方案。如您有相关技术需求,欢迎随时与我们联系~

















 2463
2463

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









