



随着测序技术的飞速发展,常规转录组测序凭借高通量、低成本、周期短的显著优势,已成为生命科学研究的“入门级”组学技术,广泛应用于农学、医学等领域,更是解析基因表达调控、挖掘功能基因的核心手段。但不少科研人常会陷入测序完成即迷茫的困境,海量数据堆积如山,却不知从何下手筛选关键信息。今天这篇攻略将从实验设计到关键基因筛选,结合经典可视化结果解读拆解,帮您搭建研究框架,快速将数据转化为科研成果。
一、转录组研究实验设计思路
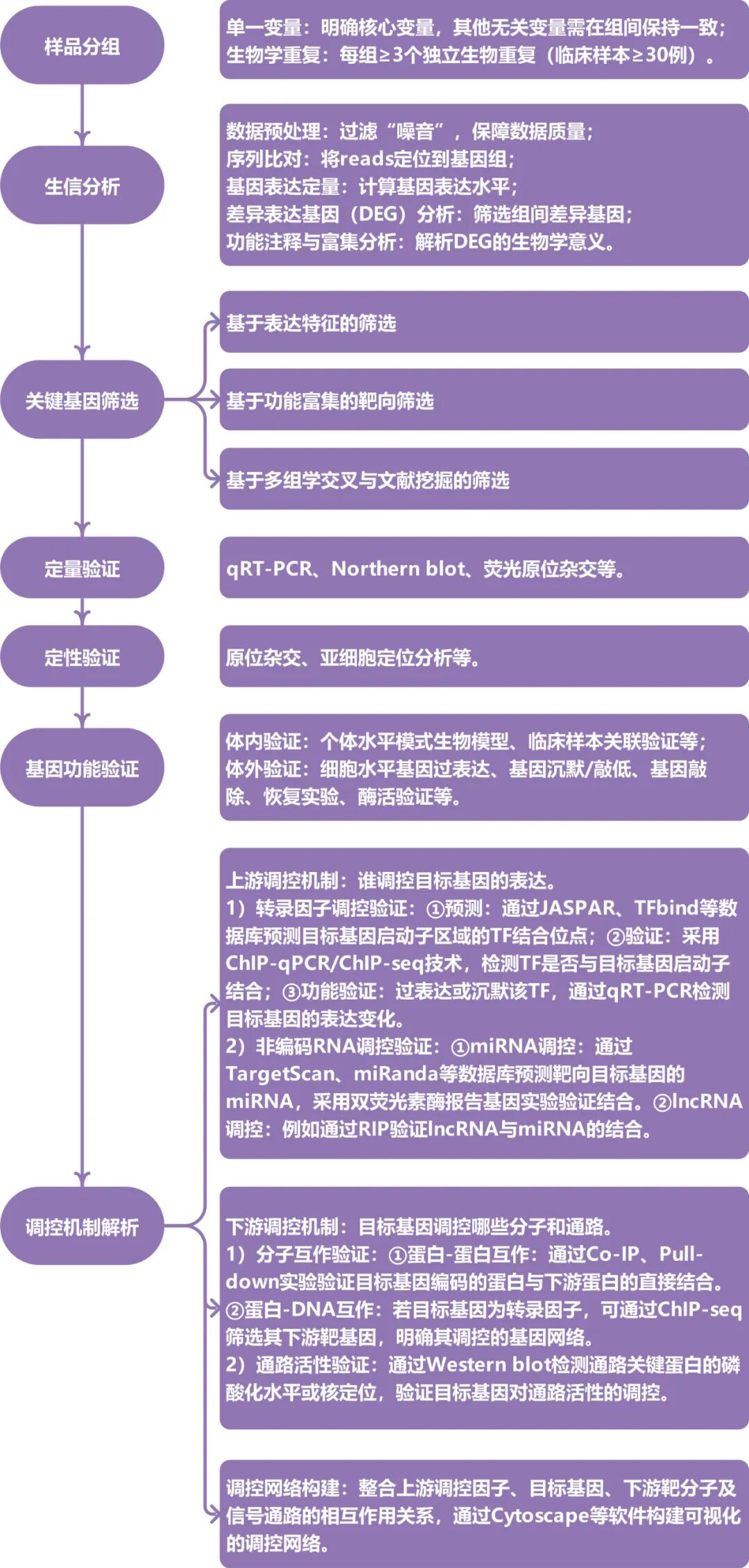
二、如何筛选关键基因
(一)差异表达基因筛选:聚焦关键基因“候选池”
差异表达基因是关键基因的“候选池”,核心是识别处理组与对照组(或不同表型组)间表达量显著差异的基因,可以通过统计显著性阈值筛选,进而结合具体功能确定关键基因。
主要筛选逻辑为:
1)若研究前期已明确关注的目标基因集,可直接定向筛选该集合内基因的差异表达情况;若未预设目标基因,则需基于差异倍数(Fold Change)和统计显著性(p值)进行排序,重点筛选极显著差异且表达倍数变化较大的基因,;
2)样本间生物学重复一致性高;
3)结合基因功能注释。
4)低表达丰度基因易受系统噪音干扰,其表达水平的可靠性较低。建议优先选择表达丰度处于中等水平的基因作为候选。
可视化展示主要有以下几种形式:
01.PCA分析:全景式观察样本异同。是否同组聚类,不同组分离,说明结果可靠且确实显著改变了基因表达模式。
拿到转录组数据,先看PCA图准没错!它是主成分分析的可视化结果,核心作用是评估样本间的整体相似性与差异,帮你快速判断实验设计的可靠性。横轴(PC1)和纵轴(PC2)分别代表第一、第二主成分。图中每个点代表一个样本,距离越近的样本,基因表达模式越相似。理想状态下,同一处理组的生物学重复样本会紧密聚集在一起(比如对照组2个重复点聚成一团),而不同处理组的样本会明显分离,这说明实验重复性好、处理因素有效。如果某一重复样本偏离同组其他样本过远,可能是样本采集污染、测序误差或操作失误导致,需进一步溯源验证,必要时剔除该样本。
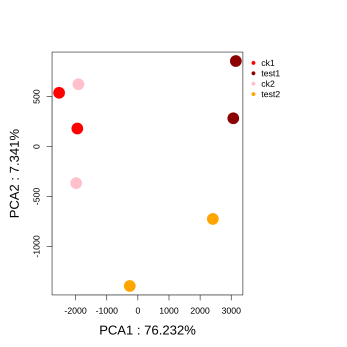
根据差异表达的基因在各个样本中的FPKM 的概念对每一个差异表达基因进行定量,通过FPKM计算样本的PCA分析。
02.火山图:差异表达基因的筛选利器。直观展示差异基因的分布,重点关注“右上/左上”区域的显著差异基因。
火山图是筛选差异表达基因最常见的结果图,能同时展示基因表达的变化倍数和统计显著性,让显著差异基因一目了然。横轴为log2(Fold Change),代表处理组与对照组的基因表达变化倍数——正值表示基因上调,负值表示下调,绝对值越大,变化幅度越显著;纵轴为-log10(PValue),表示差异显著性水平(数值越大,P值越小,显著性越高)。部分图会标注基因名称,优先关注“火山”顶端或边缘的基因——这些基因要么变化倍数极大,要么统计显著性极高,往往是后续功能验证的核心候选基因。
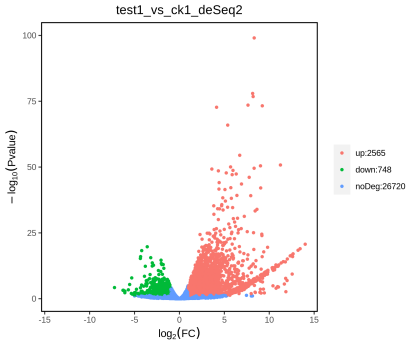
各组样本差异表达分析的火山图。
红色点(up):上调差异表达基因,共2565个;
绿色点(down):下调差异表达基因,共748个;
蓝色点(noDeg):非差异表达基因,共26720个(表达变化或显著性未达阈值)。
03.热图:基因表达模式的全景地图。对Top差异基因进行表达量聚类,验证“处理组内样本聚集、组间分离”的规律。
热图主体是颜色矩阵,每行代表一个基因,每列代表一个样本(部分图会调换行列)。颜色深浅对应基因表达量高低,常见红色表示高表达、蓝色表示低表达,具体需参考右侧图例的数值范围。热图顶部(样本聚类)和左侧(基因聚类)的树形结构是层次聚类结果。样本聚类可验证分组合理性——同组样本应聚在一起,若出现跨组聚类,需排查样本质量;基因聚类能筛选表达模式相似的基因群,这类基因往往可能参与同一生物学过程或通路。
热图的核心价值在于其模式识别能力。通过颜色分布的空间排列,可迅速识别出共表达基因簇(co-expressed gene clusters)、样本间的聚类关系以及潜在的生物学分组趋势。尤其在差异表达分析后,热图能够有效揭示在特定条件下协同上调或下调的基因群体,辅助发现关键调控通路或功能模块,并识别可能的异常表达事件或离群样本。
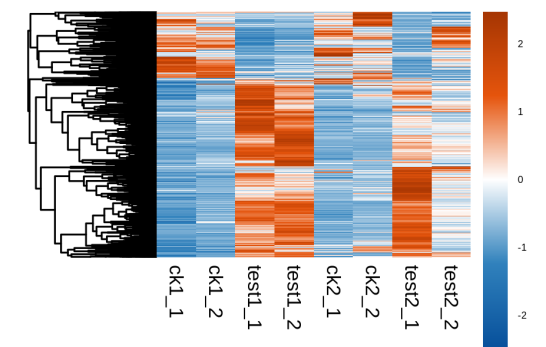
选取差异基因,对这些基因在各样本中的表达情况进行heatMap分析。
04.韦恩图:多组差异基因的交集点。通过韦恩图展示差异基因的重叠情况,锁定“共同差异基因”或“特异性差异基因”。
当需要比较多组实验(如不同处理时间、不同剂量)的差异基因集合时,韦恩图是最佳选择,用图形重叠直观展示基因集合的共有与特有关系。每个圆圈代表一组差异基因集合,圆圈的颜色或标签会标注组别信息。圆圈内的数字代表该组特有的差异基因数,而圆圈重叠区域的数字则是多组共有的差异基因数。例如,三个圆圈的交集代表在三种处理条件下均显著差异表达的基因,这类基因可能是调控核心通路的“关键节点”;而某一圆圈独有的区域则代表该处理特有的响应基因,反映处理的特异性效应。韦恩图通常用于2-4组比较,组数过多时会显得杂乱,可改用upset图展示交集关系。
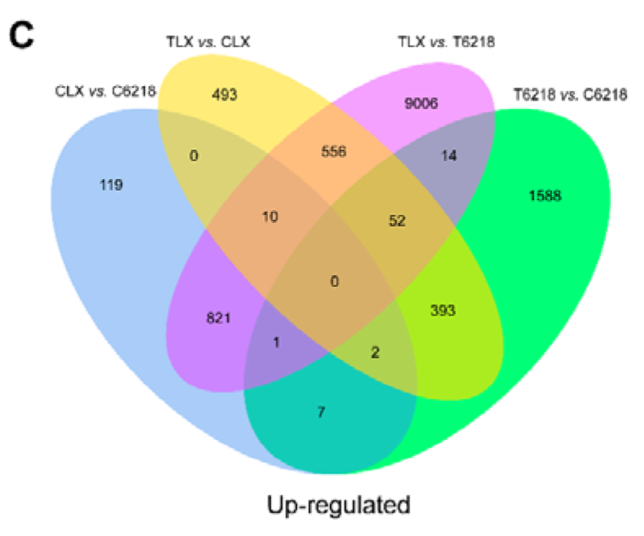
图 不同比较中上调的DEG的维恩图(Wang et al., 2022)。
05.柱状图:表达量差异结果的“直观标尺”。定量展示不同组别中的差异情况。
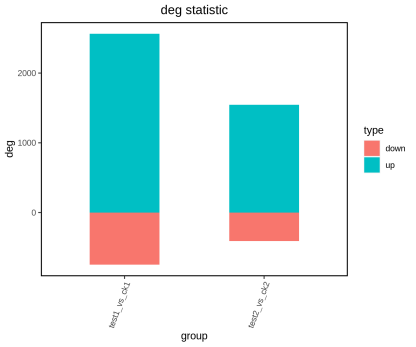
对各组差异基因进行汇总统计。
(二)功能注释与富集分析:锁定“功能相关”基因
通过富集分析将差异基因映射到具体生物学过程,筛选与研究目标(如疾病发生、胁迫响应)相关的基因。
筛选逻辑主要为:优先选择同时富集于多个功能条目且表达倍数高的基因,例如在肿瘤研究中,同时富集于“细胞增殖”和“血管生成”的差异基因更可能是关键驱动基因。
01.GO富集
分为生物学过程(BP,如“细胞凋亡”)、细胞组分(CC,如“细胞膜”)、分子功能(MF,如“蛋白激酶活性”),聚焦与研究表型直接相关的BP条目。打开GO富集分析结果文件,定位至表格中的“Term”列,通过研究方向相关的关键词(如基因功能、生物学过程等)逐一进行筛选。筛选标准通常以pvalue < 0.05作为显著富集的判定依据,符合该条件的GO Term需重点关注。在同一表格中,可直接查看所有注释到显著富集GO Term的差异基因详细信息。
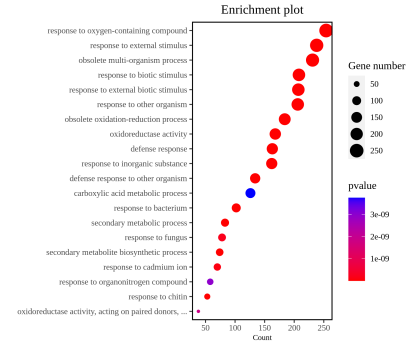
GO功能聚类气泡图展示。纵坐标是GO Term名称,横坐标是对应GO Term中检出的基因占背景基因的个数,颜色代表显著性pvalue。
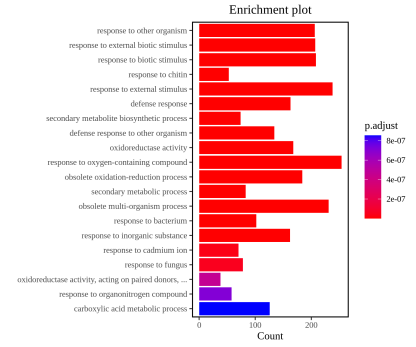
GO功能聚类bar图展示。纵坐标是GO Term名称,横坐标是对应GO Term中检出的基因个数,颜色代表显著性p.adjust。
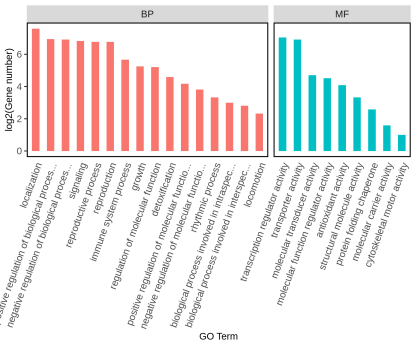
GO功能聚类wego图展示。横坐标坐标是GO Term名称,纵坐标是对应GO Term中检出的基因个数,取对数。根据GO分成了3大类,分别是分子功能(Molecular Function,MF)、细胞组分(Cellular Component,CC)、生物过程(Biological Process,BP)。
02.KEGG富集
映射到信号通路(如“PI3K-Akt通路”“MAPK通路”),筛选通路中的“关键节点基因”(如通路中的激酶、转录因子)。打开KEGG通路富集分析结果文件,找到表格中的“pathway”列,使用与研究主题相关的通路关键词进行逐一筛选。筛选标准同GO富集分析一致,以pvalue < 0.05作为显著富集通路的判定标准,此类通路需重点分析。针对筛选出的重点通路,参照上述GO富集结果的筛选方法,即可在表格中提取到所有注释到目标通路的显著差异基因信息。
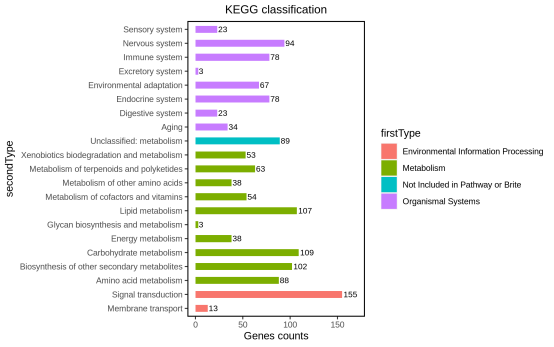
KEGG Pathway主要划分为7类:分别为Metabolism,Genetic information Processing等。其中每类又分为二、三、四级子条目。功能聚类分类图课展示pathway所属一级条目和二级条目的情况。
03.GSEA分析
若差异基因整体富集不显著,可通过基因集富集分析(GSEA)挖掘“整体趋势一致”的功能基因集。常规富集分析只关注显著差异基因,而GSEA能挖掘非显著差异基因的集体效应,更全面揭示生物学功能变化,尤其适用于差异基因较少的情况。
传统的富集分析(如GO/KEGG富集)通常需要先设定一个严格的阈值筛选差异基因,再对筛选后的基因进行功能富集。但这种方式很容易遗漏那些“单个差异不显著,但群体协同作用关键”的基因。生物调控往往是层层递进、协同作用的。想象一个通路:上游基因A的细微上调,可能通过级联效应导致下游基因E产生剧烈变化。此时,通路中大部分基因变化可能有序但微弱,无法达到严格的差异阈值。最终,只有基因E被捕获为“差异基因”,导致整个通路在传统分析中被判定为“不显著”,成为漏网之鱼。
而GSEA分析的核心优势,就在于它无需预先筛选差异基因,直接基于所有基因的表达谱进行分析,关注基因集的整体协同变化趋势。 它不要求每个基因都发生显著变化,而是寻找整个基因集成员在排序列表中的分布是否偏向于表型的某一端(如高表达或低表达),可以精准捕捉那些包含大量微弱但方向一致变化基因的重要通路,揭示传统方法可能忽略的部分。其研究思路如下:
(1)计算基因影响力指标:评估每个基因对表型差异的贡献程度。分组型样本(如对照vs处理)常用Signal2Noise,值越大表示该基因在A组(如处理组)表达越高(或负值表示在B组高)。连续型样本(如时间序列、剂量效应)可用基因表达与表型的皮尔逊相关系数。
(2)基因排序:将所有基因按照计算出的指标值从大到小进行排序。排序列表的最顶端代表对表型(如A组高表达)贡献最大的基因,最底端代表对相反表型(如B组高表达)贡献最大的基因。
(3)基因集注释:选择感兴趣的基因集数据库,将排序列表中的基因映射到这些功能通路/基因集上。
(4)计算富集分数(Enrichment Score, ES):针对关注的某个基因集(如“细胞凋亡通路基因集”),看排序后的基因列表,如果遇到该基因集中的基因,就根据其在列表中的位置增加ES;如果遇到非该基因集的基因,则减少ES。这个过程中,位于列表顶端(显著上调)或底端(显著下调)的基因集基因,会为ES贡献更大的权重。
(5)显著性检验:计算出的ES值需要评估其统计学意义。通过置换检验来验证富集结果的可靠性,多次随机打乱样本的分组标签,重新计算ES,最终得到实际ES的P值和FDR值,确保结果的科学性。
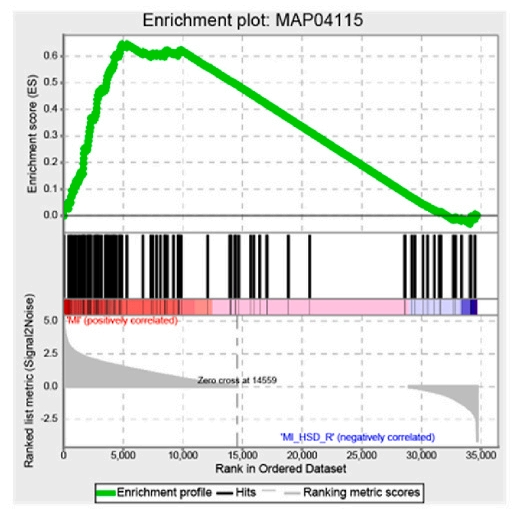
图 人类(B)和小鼠(C)基因共表达网络模块(Yao et al., 2022)。
1)上方为富集得分图:X轴是排序后的基因(从左到右:对表型贡献由大到小),Y轴是运行过程中累计的富集得分。折线走势上升段表示遇到目标基因集成员(加分),下降段表示遇到非成员(减分)。最高峰(或最低谷)对应的Y值即为该基因集的ES值。如峰出现在图左侧(高排序位置)对应正ES(顶部富集),峰出现在图右侧(低排序位置)对应负ES(底部富集)。2)中间为条形码图:黑色竖线标记排序列表中属于该基因集的基因位置。3)下方为指标排序分布图 :曲线:显示每个基因的排序指标值沿排序列表的变化。虚线:通常标记指标值为0的位置。从左到右,曲线从高正值(A组高表达/正相关)逐渐变化到高负值(B组高表达/负相关)。结合富集得分图,可直观看出显著富集的基因集成员主要分布在哪个表达/相关区间。
(三)基因共表达网络分析:挖掘“调控核心”基因
如果在不同的比较组中,差异基因显著富集的通路途径都不一致,难以逐个去筛选出关键分子,可以考虑加权基因共表达网络分析(WGCNA,Weighted Correlation Network Analysis)。WGCNA是一种基于基因表达数据构建加权共表达网络的生物信息学方法。它通过将基因间的表达相关性转化为权重,构建起类似社会网络的基因互作网络。在这个网络中,每个基因是一个节”,基因间的共表达强度是连接边的权重。随后,通过聚类分析将表达模式相似的基因划分为一个个模块,再挖掘模块与表型的关联,最终锁定关键模块和核心基因,能够高效鉴定出具有高度协同表达特征的基因模块(即基因集),并进一步结合基因模块的内部连接强度(内连性)及模块与目标表型性状的关联程度,实现候选分子筛选。其在解析复杂数据模式方面具备显著优势,尤其适用于多样本、高维度的基因表达数据研究。从样本量要求来看,当样本数量不少于15个时,可满足该分析方法的基本统计效力需求,可以开展WGCNA分析。其研究思路如下:
(1)数据预处理:首先需要对原始表达数据进行过滤,剔除表达量极低、变异度极小的基因(这类基因通常无生物学意义),同时处理缺失值和异常样本。经过预处理后,保留的基因数据集会更具分析价值。
(2)构建加权共表达网络:是WGCNA的核心步骤,关键在于确定软阈值。传统共表达分析常用硬阈值(如相关系数>0.8即为显著共表达),容易丢失弱但真实的基因关联;而WGCNA采用软阈值,通过幂函数将基因间的皮尔逊相关系数转化为权重,使得网络满足“无标度网络”特性(即少数基因与大量基因相连,符合生物网络的普遍规律)。当软阈值确定后,即可计算基因间的权重矩阵,并转化为拓扑重叠矩阵,用于衡量基因间的相似度,拓扑重叠度越高,基因的共表达模式越相似。
(3)模块识别与合并:基于拓扑重叠矩阵,WGCNA会通过层次聚类将基因划分为不同的模块,每个模块用不同颜色标记。通常会设置模块合并阈值,将表达模式高度相似的模块进行合并,避免模块过多导致分析复杂。每个模块都有一个特征向量基因,它代表了整个模块的核心表达模式,后续的表型关联分析主要围绕特征向量基因展开。
(4)模块-表型关联与核心基因筛选:将每个模块的特征向量基因与研究的表型数据。进行相关性分析,筛选出与目标表型显著相关的“关键模块”。在关键模块中,再通过计算基因显著性(GS,基因与表型的相关性)和模块成员性(MM,基因与模块特征向量的相关性),筛选出GS和MM均较高的基因,这类基因就是模块中的“枢纽基因(Hub Gene)”,极有可能是调控表型的核心基因。
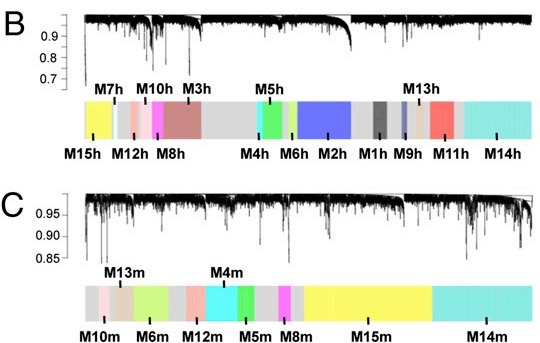
图 人类(B)和小鼠(C)基因共表达网络模块(Miller et al., 2010)。
树状图结构纵轴:基因间距离(Distance = 1 - TO值),TO越低则距离越大,横轴:基因按共表达相似性排列模块划分结果,通过动态树切割法识别显著分支点,图示底部色块标记不同模块,显著重叠的模块分配相同标签(如M1h, M2h等)。
(四)跨数据集验证:提升“可靠性”基因
单一数据集的结果可能存在批次效应或样本偏差,可以通过跨数据集验证筛选“稳定差异”的关键基因。
1、公共数据集验证:从GEO、TCGA等数据库下载同类型数据集(如相同疾病的不同队列、相同胁迫的不同品种),验证候选基因在不同数据集中的差异趋势一致性。
2、物种间保守性验证:若研究模式生物(如小鼠、拟南芥),可通过同源基因分析验证候选基因在人类或其他物种中的功能保守性。
小结
看完以上解读,是不是对转录组图表更有底气了?最后分享一个核心原则:图表解读永远服务于生物学问题,先明确自己的研究目的(如筛选差异基因、挖掘通路、关联性状),再针对性聚焦图表的关键信息,结合实验设计推导生物学意义。无论采用“先找差异基因再看功能”,还是“先从功能出发聚焦基因”的策略,人为的深度解读和针对性数据挖掘始终是不可替代的核心环节。测序软件和分析工具能高效完成数据的筛选和注释,但无法替代研究者对生物学问题的深刻理解。例如,某基因表达量变化不显著但在通路中处于关键调控节点,或某基因在特定组织/处理时间点的特异性表达,这些隐藏信息只有结合研究背景的人为分析才能精准捕捉。
若你在转录组实验设计、数据解析或后续验证环节遇到技术瓶颈,或是有定制化的研究方案需求,欢迎随时联系我们。我们的技术团队将凭借丰富的实战经验,为你的科研项目保驾护航。祝愿每一位科研同仁都能在探索生命奥秘的道路上硕果累累,科研顺利,成果丰硕,早日在高水平期刊上发表佳作!
参考文献
[1] Wang Q, Liu X, Liu H, et al. Transcriptomic and metabolomic analysis of wheat kernels in response to the feeding of orange wheat blossom midges (Sitodiplosis mosellana) in the field[J]. Journal of Agricultural and Food Chemistry, 2022, 70(5): 1477-1493.
[2] Yao M, Lu Y, Shi L, et al. A ROS-responsive, self-immolative and self-reporting hydrogen sulfide donor with multiple biological activities for the treatment of myocardial infarction[J]. Bioactive materials, 2022, 9: 168-182.
[3] Miller J A, Horvath S, Geschwind D H. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways[J]. Proceedings of the National Academy of Sciences, 2010, 107(28): 12698-12703.

















 2031
2031

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









