




古早系列博客的补充,参考我之前的博客:https://blog.youkuaiyun.com/weixin_62528784/article/details/146487094?spm=1001.2014.3001.5502
参考:
https://prosite.expasy.org/prosuser.html#conv_de

序列比对获取不了的信息,从motif/结构域的基本特征unit上去识别(获取信息);
序列中出现的特定残基类型簇来识别,这些簇被称为模式、基序、签名或指纹。这些基序的出现是因为蛋白质特定区域(可能很重要)的结构有特定要求,例如,对于它们的结合特性或酶活性。这些要求对蛋白质序列中这些有限但重要的部分(在大小上)的进化施加了非常严格的限制。
我们分析结构域的,无论如何讲故事,故事的主线都得和结构、进化挂钩,
因为这个是生物的nature,是自然界的真理,
无论domain有多种多样各种阿猫阿狗的特征信息feature,我们首要关注的应该是结构structure(当然序列信息是基础,这里说的只是我们要关联功能,应该关注的是序列反映在结构层次上的信息,结构是最直接、最本质的信息);
因为domain/motif必定是以结构特征为中心的表征,必定是进化信息的体现(这是我的理解)
基序、模板或指纹的出现,是因为结合位点对蛋白序列部分进化施加了非常严格的约束。
motif的特殊性或者说是出现,正是因为进化信息的体现(所以能够反映或者说是表征)

传统的模式检测方法依赖于固定的氨基酸序列模式。例如,假设我们有一个简单的模式[RK]X[DE],其中[RK]表示赖氨酸(K)或精氨酸(R),X表示任意氨基酸,[DE]表示天冬氨酸(D)或谷氨酸(E)。
这种模式可以用来检测某些特定的磷酸化位点,但如果蛋白质序列中存在大量变异,这种简单的模式就可能无法匹配到目标序列。
——》我们需要考虑到变异情况下,即进化事件下的序列表征
例如,假设我们有以下两个蛋白质序列片段:
- 序列A:
RKXDE - 序列B:
RAXEE
对于序列A,传统模式[RK]X[DE]可以完美匹配,但对于序列B,由于X位置周围邻居的氨基酸发生了变化,传统模式无法检测到其潜在的功能。
基于权重矩阵(轮廓)的技术
基于权重矩阵(也称为轮廓,profile)的技术能够克服传统模式检测方法的局限性。权重矩阵通过考虑每个氨基酸位置的多种可能性及其权重来检测蛋白质的功能或结构域。这种方法的核心是位置特异性评分矩阵(PSSM),它为每个氨基酸位置分配一个分数,表示该位置上不同氨基酸出现的概率。
——》其实说白了就是PSSM(转录因子结合位点TFBS概念里的motif)模型,其效果要优于最原始最朴素的正则表达式;
从理论上来讲:识别某一个蛋白质结构域,比如说要识别Krab结构域,
我可以使用这个结构域的正则表达式,
但是对于退化的、非典型的,总之是由于非典型进化事件导致的一些突变、变异,实际上正则表达式的检测detect能力就弱得多了;
我们定义1个domain/motif,从逻辑上来看,是结构决定事件的,所以本质上我们应该看不同的、相似的结构能否导向同样的生物学功能、动力学,
从序列到结构的角度分析,我们当然可以扩充此处的序列可能性,
但是正则表达式的扩充相对困难点(技术表示上),
一个位置权重矩阵,其实就是拟合了1个概念分布(频率分布),虽然也是缝缝补补的逻辑,但是单单从技术实现的角度上讲,还是要比单纯正则表达式容易的。
在蛋白质结构域识别中,正则表达式(Regular Expression)和位置权重矩阵(Position Weight Matrix, PWM)是两种不同但互补的方法。以 C2H2型锌指蛋白(Zinc Finger Protein, ZFP) 的结构域为例,
1. 正则表达式(Regular Expression)
原理
正则表达式通过定义特定字符模式匹配序列。对于 C2H2锌指结构域,其保守特征是:
- 两个半胱氨酸(C)结合一个锌离子,随后两个组氨酸(H)结合另一个锌离子。
- 典型模式为:
C-X2-4-C-X12-H-X3-5-H,其中X表示任意氨基酸,数字表示间隔长度。
正则表达式示例:
C.{2,4}C.{12}H.{3,5}H
特点
- 优势:
- 简单直观,计算速度快。
- 适合检测高度保守的基序(如严格的间隔长度和关键残基)。
- 局限性:
- 无法容忍氨基酸替换或间隔长度的微小变化(如
X12变为X11或X13)。 - 忽略中间位置的氨基酸偏好(如疏水性或极性要求)。
- 无法容忍氨基酸替换或间隔长度的微小变化(如
示例匹配失败:
若某序列中第二个半胱氨酸(C)后的间隔为 X11,或某位点存在保守的疏水残基(如亮氨酸)被替换为丙氨酸,正则表达式将无法识别。
2. 位置权重矩阵(PWM)
原理
PWM 基于统计学模型,为每个位置赋予不同氨基酸的权重分数。以 C2H2 锌指为例:
- 收集大量已知 C2H2 结构域的序列。
- 统计每个位置上氨基酸的分布频率。
- 构建矩阵,计算每个位置的氨基酸得分(如对数几率比)。
PWM 匹配流程:
- 滑动窗口遍历序列,计算每个窗口的总分。
- 若总分超过阈值,判定为匹配。
特点
- 优势:
- 容忍氨基酸替换(如某位置允许丙氨酸或丝氨酸)。
- 捕捉中间位置的保守偏好(如疏水残基的统计倾向)。
- 可识别间隔长度的轻微变化(通过动态规划或窗口滑动)。
- 局限性:
- 依赖训练数据集的质量和多样性。
- 计算复杂度高于正则表达式。
示例匹配成功:
若某序列的第二个半胱氨酸(C)后间隔为 X11,但其他位置氨基酸符合统计偏好(如疏水残基),PWM 仍可能识别为 C2H2 结构域。
3. 正则表达式 vs PWM 的联系
- 共同目标:检测序列中的功能或结构域。
- 互补性:
- 正则表达式适合快速筛选高度保守的基序。
- PWM 适合检测复杂、变异性高的基序。
- 联合应用:
- 先用正则表达式粗筛候选序列,再用 PWM 精细验证。
4. 实际应用中的功效对比
C2H2 ZFP 结构域检测案例
- 正则表达式:
- 能快速找到严格符合
C-X2-4-C-X12-H-X3-5-H模式的序列。 - 漏检率较高(如变异或长度偏离的锌指)。
- 能快速找到严格符合
- PWM:
- 检测到更多远缘同源结构域(如间隔长度波动或保守替换)。
- 假阳性率可能略高(需结合阈值优化)。
总结
| 方法 | 适用场景 | 优势 | 局限性 |
|---|---|---|---|
| 正则表达式 | 高度保守、严格模式的快速检测 | 简单、高效 | 无法容忍变异 |
| 位置权重矩阵 | 复杂、变异大的结构域精细化识别 | 灵活、统计驱动 | 依赖数据、计算复杂 |
在 C2H2 ZFP 检测中,PWM 更适用于发现进化距离较远或序列变异的锌指结构域,而正则表达式适合初步筛选经典模式。两者结合可提升检测的敏感性和特异性。


一,模式pattern更新:


我们关注pattern,也就是对domain的研究,也应该关注下面方面:




上面的说法其实很简单:
意思就是我们只想要匹配上面的那3条序列,我们最初的A-T-H-[D or E]模式实际上太短了,匹配的假阳性大(意思就是说会匹配到很多其他不是这3条序列的序列,说白了就是这条规则太简单了);
如果我们进一步扩展这个4字符的核心模式的话,我们当然能够得到完全匹配的扩展模式(就是上面提到的过拟合的模式)。对,就是过拟合了!
而且这种过拟合是没有意义的。至少是没有生物学意义的

数据里搜索的思维,对于同一条序列中出现的多种motif/结构域序列,也是要求不重叠的(当然我们知道如果依据滑动窗口、双指针那一套去搞,step size为1,其实能够找到更多,但是结构域重叠可能没有显式的生物学意义,这一点其实和原核或者真核生物中的ORF scanning不太一样,ORF其实是可以有嵌套ORF的,开放阅读框架,也就是基因结构识别)。
**另外我们重点要关注的是profile.txt文件:
**profile.txt 文件提供了profile结构和相应的基序搜索方法的技术描述(主要是提供了技术方法描述)。

另外profile(也就是位置权重矩阵那一套)实际上能够检测得更多pattern,道理很简单,分配权重的氨基酸残基多,组合就多

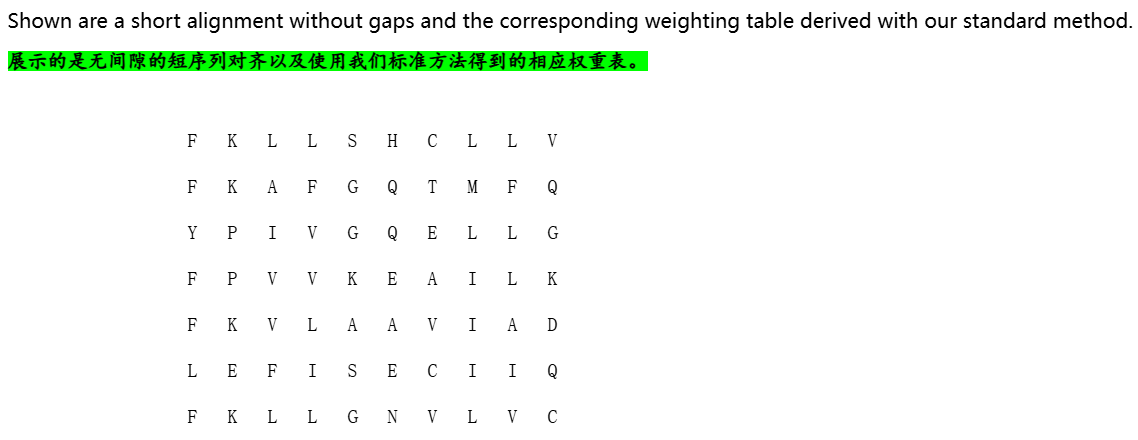


这个PWM其实就容易理解:
比如说长度为10的这么个窗口序列,第1个位置F score最高,最可能出现
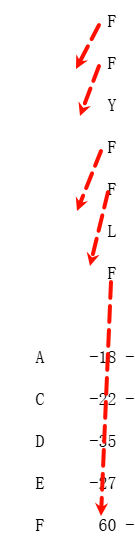
第2个位置是k:
同理以此类推

















 1525
1525

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









