







一,背景:
PROSITE:

PROSITE数据库收集了生物学有显著意义的蛋白质位点和序列模式,并能根据这些位点和模式快速和可靠地鉴别一个未知功能的蛋白质序列应该属于哪一个蛋白质家族。有的情况下,某个蛋白质与已知功能蛋白质的整体序列相似性很低,但由于功能的需要保留了与功能密切相关的序列模式,这样就可能通过PROSITE的搜索找到隐含的功能motif,因此是序列分析的有效工具。PROSITE中涉及的序列模式包括酶的催化位点、配体结合位点、与金属离子结合的残基、二硫键的半胱氨酸、与小分子或其它蛋白质结合的区域等;除了序列模式之外,PROSITE还包括由多序列比对构建的profile,能更敏感地发现序列与profile的相似性。PROSITE的主页上提供各种相关检索服务。
1,PROSITE中的Pattern syntax rules
Pattern syntax rules # 模式语法规则
1.The standard IUPAC one-letter codes for the amino acids are used.
# 使用氨基酸的标准IUPAC单字母编码
2.The symbol `x' is used for a position where any amino acid is accepted.
# 符号“x”表示任何氨基酸都可以被接受的位置,代表单个任意氨基酸
3.Ambiguities are indicated by listing the acceptable amino acids for a given position, between square brackets `[ ]'. For example: [ALT] stands for Ala or Leu or Thr.
# 通过在方括号‘[]’之间列出给定位置的可接受的氨基酸来表示歧义。例如:[ALT]代表Ala、Leu或Thr。其实就是Or匹配,在方括号中的aa单字母
4.Ambiguities are also indicated by listing between a pair of curly brackets `{ }' the amino acids that are not accepted at a given position. For example: {AM} stands for any amino acid except Ala and Met.
# 通过在一对花括号“{}”之间列出在给定位置不被接受的氨基酸,也可以表示歧义。例如:{AM}代表除Ala和Met以外的任何氨基酸。同上
5.Each element in a pattern is separated from its neighbor by a `-'.
# 模式中的每个元素通过‘ -’与相邻元素分隔
6.Repetition of an element of the pattern can be indicated by following that element with a numerical value or, if it is a gap ('x'), by a numerical range between parentheses.
Examples:
x(3) corresponds to x-x-x
x(() 2,4) corresponds to x-x or x-x-x or x-x-x-x
A(3) corresponds to A-A-A
Note: You can only use a range with 'x', i.e. A(2,4) is not a valid pattern element.
# 模式中某个元素的重复可以通过在该元素后面加上一个数值来表示,如果元素是一个间隙('x'),则可以通过括号之间的数值范围来表示。
# 例子:
# X(3)对应X - X - X
# X(() 2,4)对应X - X或X - X - X或X - X - X - X
# A(3)对应A-A-A
# 注意:您只能使用带有‘x’的范围,即A(2,4)不是有效的模式元素。
7.When a pattern is restricted to either the N- or C-terminal of a sequence, that pattern either starts with a `<' symbol or respectively ends with a `>' symbol. In some rare cases (e.g. PS00267 or PS00539), '>' can also occur inside square brackets for the C-terminal element. 'F-[GSTV]-P-R-L-[G>]' means that either 'F-[GSTV]-P-R-L-G' or 'F-[GSTV]-P-R-L>' are considered.
# 当一个模式被限制在序列的N端或c端时,该模式要么以‘ <’符号开始,要么分别以‘ >’符号结束。在一些罕见的情况下(例如PS00267或PS00539), ‘>’也可以出现在c端元素的方括号内。“F - [GSTV] -P-R-L > [G]意味着“F - [GSTV] -P-R-L-G”或者“F - [GSTV] -P-R-L >”。
Information above comes from http://www.hpa-bioinfotools.org.uk/ps_scan/PS_SCAN_PATTERN_SYNTAX.html.
其实关键就在下面的几个匹配符
[]: sets of allowed amino acids
{}: sets of disallowed amino acids
-: separator between pattern elements
(x) and (x,y): length ranges
<,>: anchor to N- or C-terminus
.: end of pattern.
2,联系正则表达式/grep:
二,PROSITE pattern scanner 手写模式匹配
本质上就是写个正则表达式,关键在于正则表达式的符号
[]: sets of allowed amino acids
{}: sets of disallowed amino acids
-: separator between pattern elements
(x) and (x,y): length ranges
<,>: anchor to N- or C-terminus
.: end of pattern.
 上面的是常规的正则表达式的写法,没有使用re库等,下面的是使用re正则表达式的匹配库,实际上是类似的。
上面的是常规的正则表达式的写法,没有使用re库等,下面的是使用re正则表达式的匹配库,实际上是类似的。

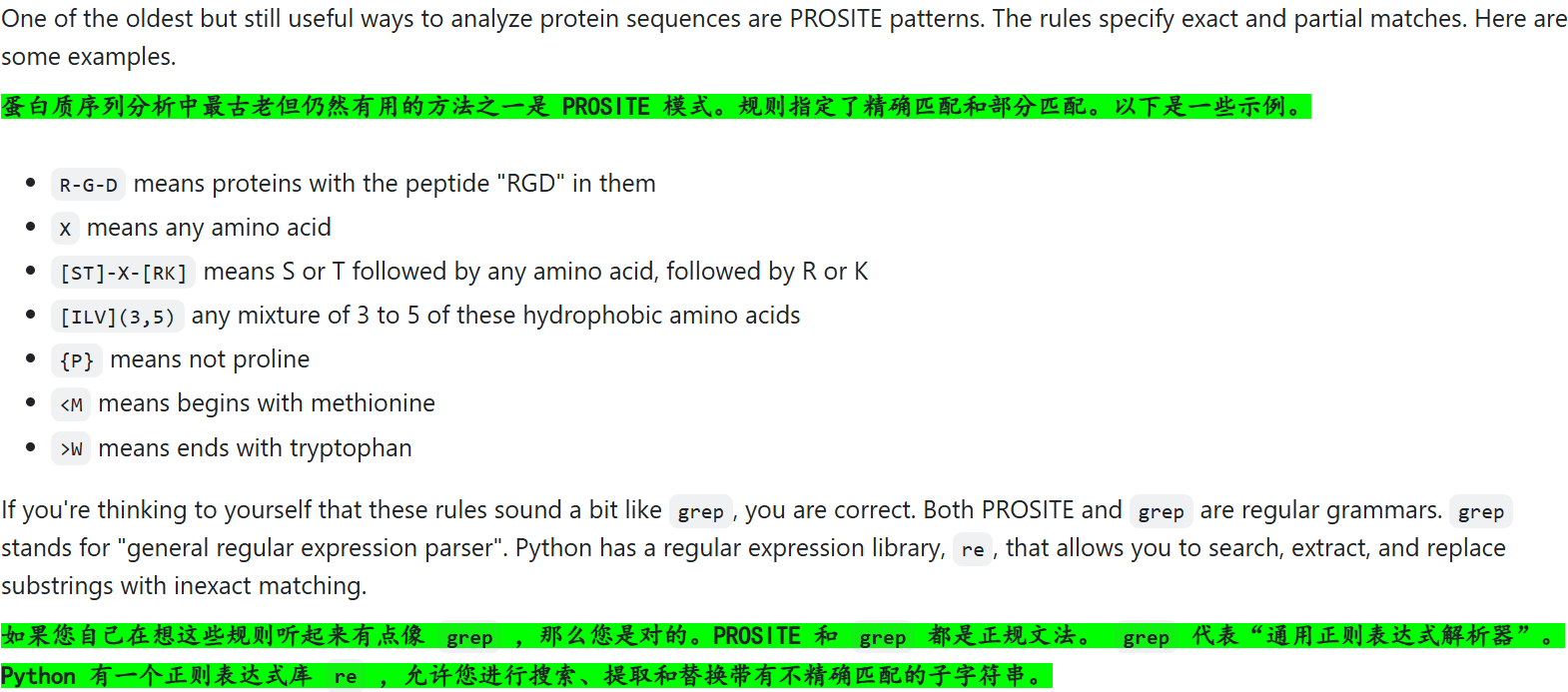
但是事实上,我们能够看到,常规如果不使用re库等来匹配正则表达式的话,对于复杂的序列模式其实是表征很困难的;
所以我们需要使用re等正则表达式库:
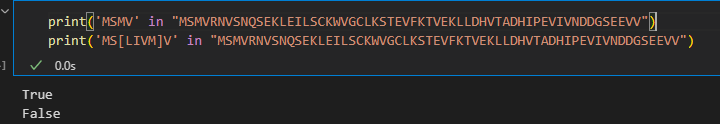
主要就是search函数以及finditer、findall函数,
我们先拿一个简单的例子来看看:还是CTCF,
然后我们选取的PROSITE正则表达式为Rule PS00028

按理来说可以使用这个正则表达式的话,我们是能够匹配到8个这样的结构域的,
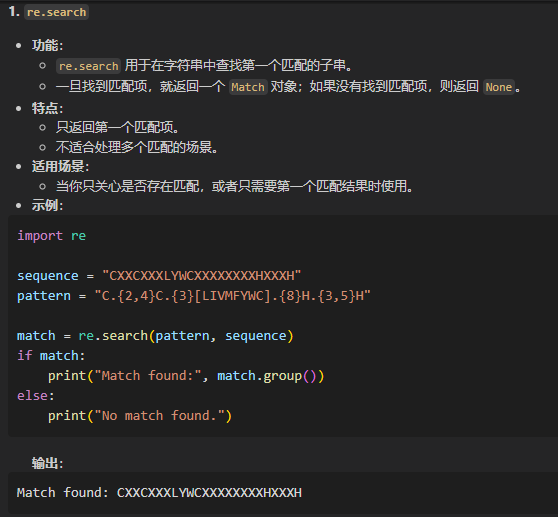
1,re.search
re.search("C.{2,4}C.{3}[LIVMFYWC].{8}H.{3,5}H","MEGDAVEAIVEESETFIKGKERKTYQRRREGGQEEDACHLPQNQTDGGEVVQDVNSSVQMVMMEQLDPTLLQMKTEVMEGTVAPEAEAAVDDTQIITLQVVNMEEQPINIGELQLVQVPVPVTVPVATTSVEELQGAYENEVSKEGLAESEPMICHTLPLPEGFQVVKVGANGEVETLEQGELPPQEDPSWQKDPDYQPPAKKTKKTKKSKLRYTEEGKDVDVSVYDFEEEQQEGLLSEVNAEKVVGNMKPPKPTKIKKKGVKKTFQCELCSYTCPRRSNLDRHMKSHTDERPHKCHLCGRAFRTVTLLRNHLNTHTGTRPHKCPDCDMAFVTSGELVRHRRYKHTHEKPFKCSMCDYASVEVSKLKRHIRSHTGERPFQCSLCSYASRDTYKLKRHMRTHSGEKPYECYICHARFTQSGTMKMHILQKHTENVAKFHCPHCDTVIARKSDLGVHLRKQHSYIEQGKKCRYCDAVFHERYALIQHQKSHKNEKRFKCDQCDYACRQERHMIMHKRTHTGEKPYACSHCDKTFRQKQLLDMHFKRYHDPNFVPAAFVCSKCGKTFTRRNTMARHADNCAGPDGVEGENGGETKKSKRGRKRKMRSKKEDSSDSENAEPDLDDNEDEEEPAVEIEPEPEPQPVTPAPPPAKKRRGRPPGRTNQPKQNQPTAIIQVEDQNTGAIENIIVEVKKEPDAEPAEGEEEEAQPAATDAPNGDLTPEMILSMMDR")

我们发现search只返回了1个,总体还是接近的,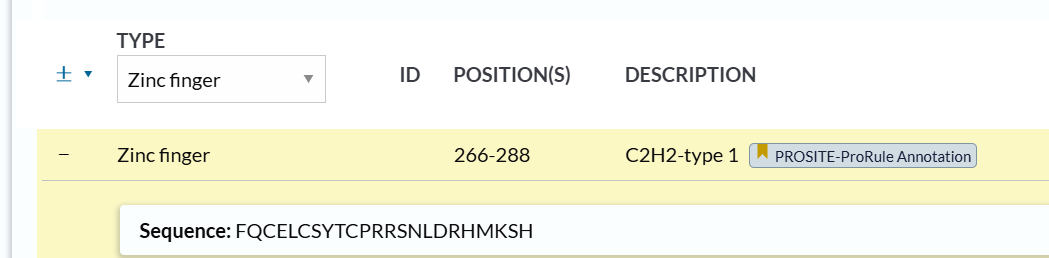
当然因为此处CTCF的注释使用的不是这个规则,所以说是接近而不是精确匹配

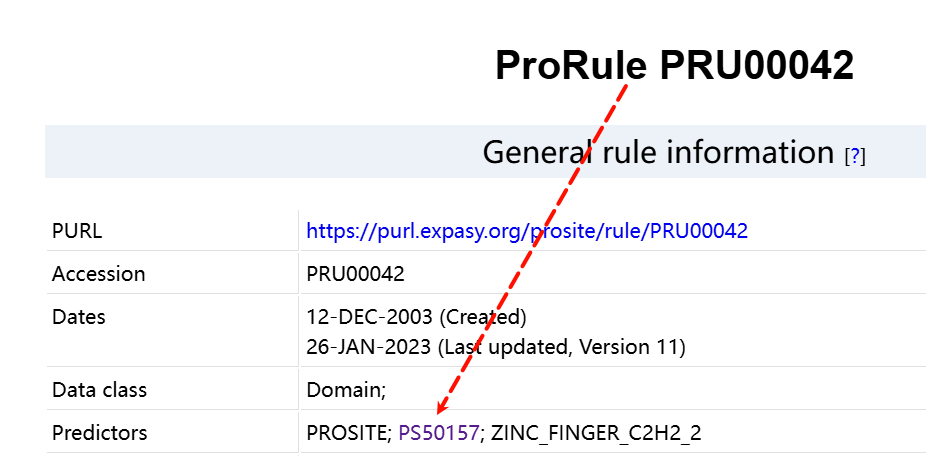
当然我们可以仔细查看一下细节:
result1 = re.search("C.{2,4}C.{3}[LIVMFYWC].{8}H.{3,5}H","MEGDAVEAIVEESETFIKGKERKTYQRRREGGQEEDACHLPQNQTDGGEVVQDVNSSVQMVMMEQLDPTLLQMKTEVMEGTVAPEAEAAVDDTQIITLQVVNMEEQPINIGELQLVQVPVPVTVPVATTSVEELQGAYENEVSKEGLAESEPMICHTLPLPEGFQVVKVGANGEVETLEQGELPPQEDPSWQKDPDYQPPAKKTKKTKKSKLRYTEEGKDVDVSVYDFEEEQQEGLLSEVNAEKVVGNMKPPKPTKIKKKGVKKTFQCELCSYTCPRRSNLDRHMKSHTDERPHKCHLCGRAFRTVTLLRNHLNTHTGTRPHKCPDCDMAFVTSGELVRHRRYKHTHEKPFKCSMCDYASVEVSKLKRHIRSHTGERPFQCSLCSYASRDTYKLKRHMRTHSGEKPYECYICHARFTQSGTMKMHILQKHTENVAKFHCPHCDTVIARKSDLGVHLRKQHSYIEQGKKCRYCDAVFHERYALIQHQKSHKNEKRFKCDQCDYACRQERHMIMHKRTHTGEKPYACSHCDKTFRQKQLLDMHFKRYHDPNFVPAAFVCSKCGKTFTRRNTMARHADNCAGPDGVEGENGGETKKSKRGRKRKMRSKKEDSSDSENAEPDLDDNEDEEEPAVEIEPEPEPQPVTPAPPPAKKRRGRPPGRTNQPKQNQPTAIIQVEDQNTGAIENIIVEVKKEPDAEPAEGEEEEAQPAATDAPNGDLTPEMILSMMDR")
print(result1)
print(result1.start(), result1.end())
print(result1.group())
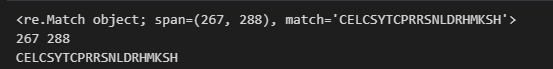
是能够返回匹配的起点、终点以及序列的
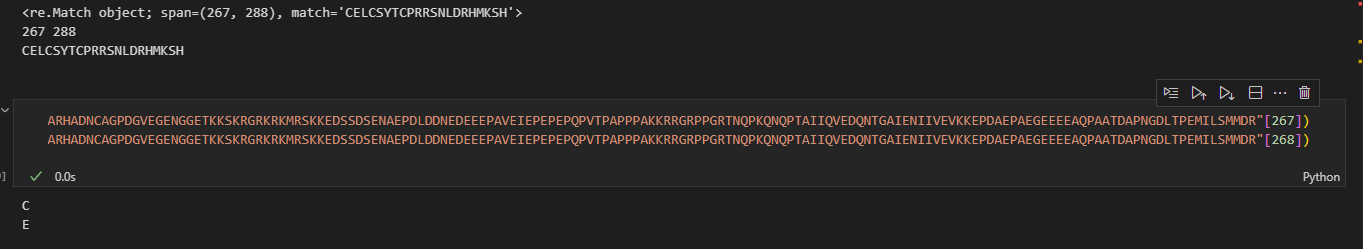
然后这里的index实际上是0-based,这一点很清楚
2,re.findall
紧接着,我们又看了findall:
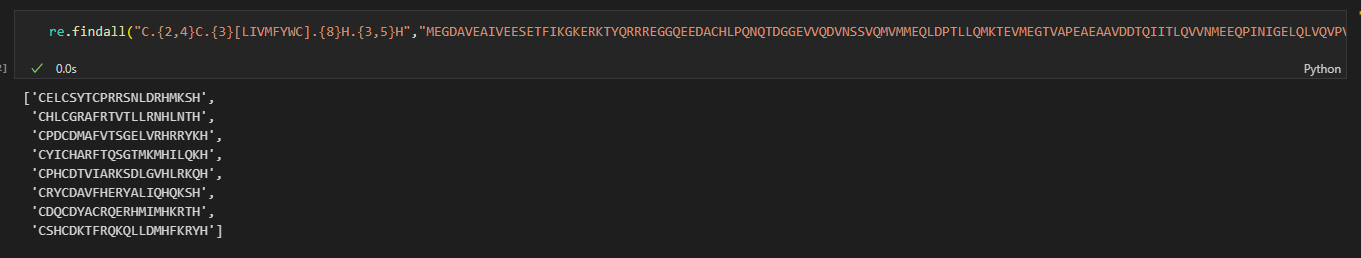
这个是直接返回结果匹配的字符串的,正好是8个,
但是因为不是re.Match对象,所以我们不能够提取出什么有用的信息
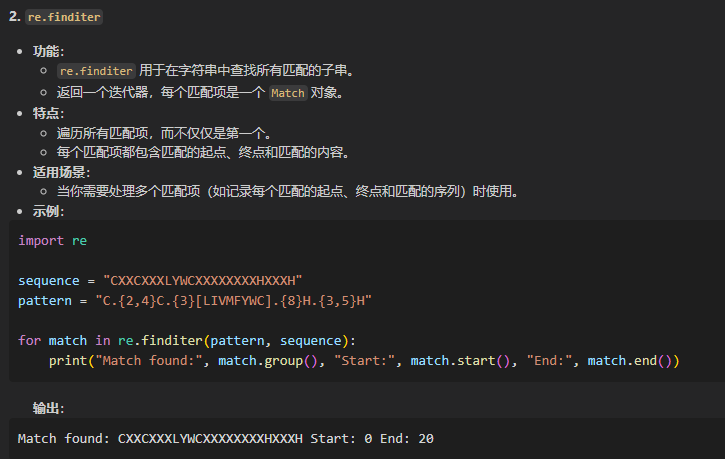
3,re.finditer
result2 = re.finditer("C.{2,4}C.{3}[LIVMFYWC].{8}H.{3,5}H","MEGDAVEAIVEESETFIKGKERKTYQRRREGGQEEDACHLPQNQTDGGEVVQDVNSSVQMVMMEQLDPTLLQMKTEVMEGTVAPEAEAAVDDTQIITLQVVNMEEQPINIGELQLVQVPVPVTVPVATTSVEELQGAYENEVSKEGLAESEPMICHTLPLPEGFQVVKVGANGEVETLEQGELPPQEDPSWQKDPDYQPPAKKTKKTKKSKLRYTEEGKDVDVSVYDFEEEQQEGLLSEVNAEKVVGNMKPPKPTKIKKKGVKKTFQCELCSYTCPRRSNLDRHMKSHTDERPHKCHLCGRAFRTVTLLRNHLNTHTGTRPHKCPDCDMAFVTSGELVRHRRYKHTHEKPFKCSMCDYASVEVSKLKRHIRSHTGERPFQCSLCSYASRDTYKLKRHMRTHSGEKPYECYICHARFTQSGTMKMHILQKHTENVAKFHCPHCDTVIARKSDLGVHLRKQHSYIEQGKKCRYCDAVFHERYALIQHQKSHKNEKRFKCDQCDYACRQERHMIMHKRTHTGEKPYACSHCDKTFRQKQLLDMHFKRYHDPNFVPAAFVCSKCGKTFTRRNTMARHADNCAGPDGVEGENGGETKKSKRGRKRKMRSKKEDSSDSENAEPDLDDNEDEEEPAVEIEPEPEPQPVTPAPPPAKKRRGRPPGRTNQPKQNQPTAIIQVEDQNTGAIENIIVEVKKEPDAEPAEGEEEEAQPAATDAPNGDLTPEMILSMMDR")
print(result2)
for match in result2:
print(match.start(), match.end(), match.group())
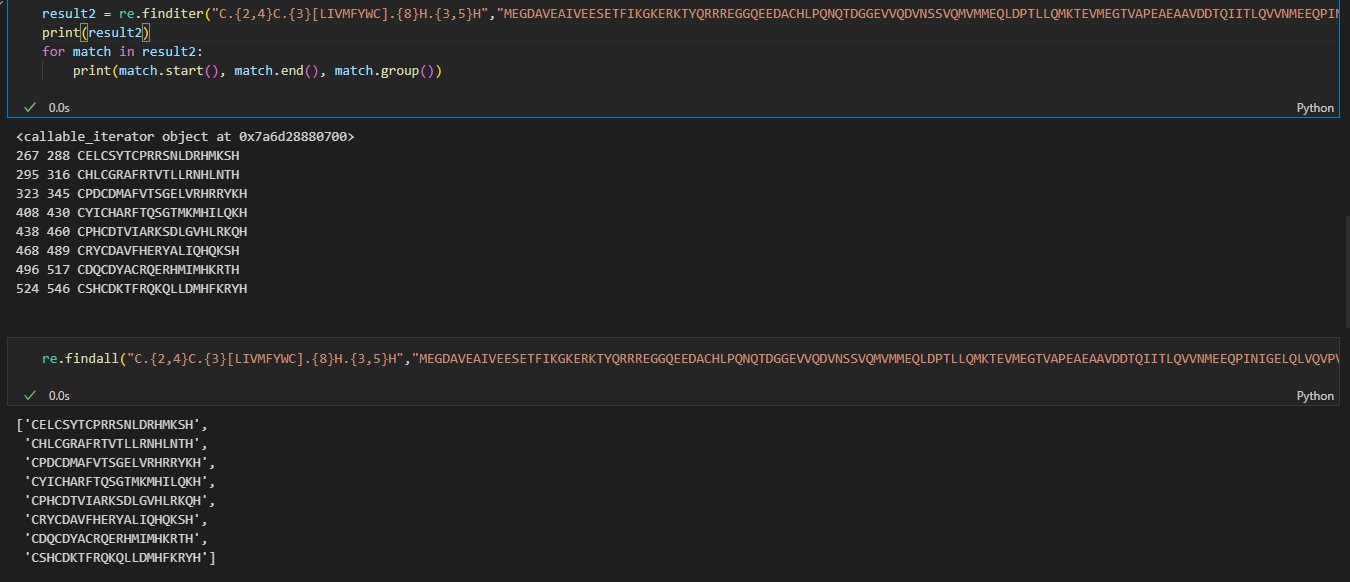
其实,我们仔细对照一下,就能够发现finditer的结果比findall更加丰富,实际上就是findall就是finditer中的group结果。
当然,这里其实还有一个小小的逻辑问题:re.finditer事实上每次匹配的时候都将上一次匹配的终点作为下一次匹配的起点,所以本质上是非重叠匹配;
我们如果严谨一点,也应该考虑一下重叠部分的匹配,那么这个时候我们其实就需要使用re.search(这个函数只寻找first匹配,所以实际上我们可以使用这个函数写一个loop,然后每次手动修改每次匹配之后起点只移动一个字符,而不是直接跳到当前匹配的结束位置);
当然,实际上我们在寻找结构域,而且还是同一个结构域的时候,尤其是在生物学背景中,我还没见过有所谓两个结构域重合在一起的情况,我想生物学中是不会这么定义的,所以我们还是使用前面的非重叠匹配即可。
当然,此处也提供重叠匹配的code实现逻辑:
import re
def find_overlapping_matches(pattern, sequence):
"""
查找所有匹配项,包括重叠的部分。
Args:
pattern: str, 正则表达式模式。
sequence: str, 要匹配的字符串。
Returns:
list: 包含所有匹配项的列表,每个匹配项是一个 (start, end, match) 元组。
"""
matches = []
start = 0
while start < len(sequence):
match = re.search(pattern, sequence[start:])
if not match:
break
# 记录匹配的起点、终点和匹配的内容
match_start = start + match.start()
match_end = start + match.end()
matches.append((match_start, match_end, match.group()))
# 从下一个字符开始继续匹配,允许重叠
start = match_start + 1
return matches
注意上面code中break的逻辑:
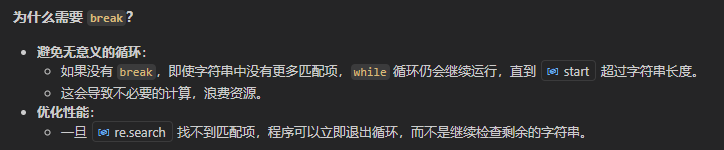
break的逻辑在于:如果当前起点的字符串序列中都没有找到第1个match匹配项,就更不可能会有重叠等index更后面的match项了。
综上,我们的经验是:
我们在程序(非重叠匹配问题,该背景中)中使用re.finditr,
如果是重叠问题,我们会使用re.search函数手动编写调整循环匹配起点的程序,自由度更大
三,issue(小插曲):
实际上我依据这个小idea写了个小py程序(作为1个小模块),
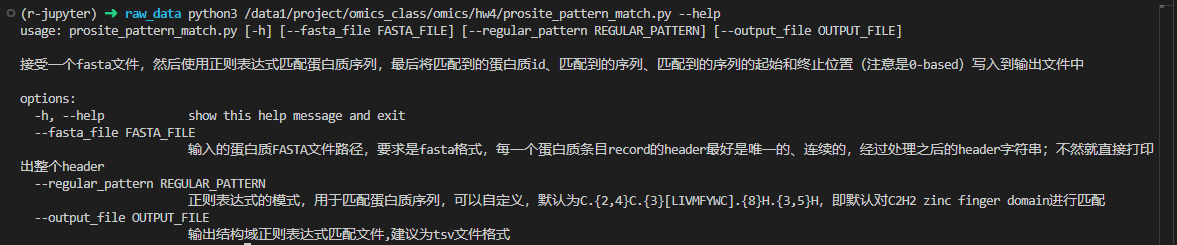

python3 prosite_pattern_match.py --fasta_file /lustre/share/class/BIO8402/hw4/ws_215.protein.fa --output_file c2h2_zf_ws_215.stv
但是运行的时候报错了:
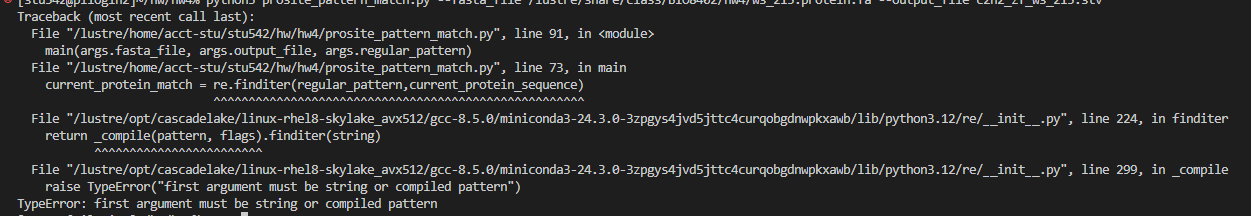
报错信息很明显,其实就是finditer中的正则表达式的匹配项参数没有正确传入,
事实上,正是因为我在main主函数中提供了该参数的默认值,我就没在调用函数中传入该参数了,我就犯了一个很常见的错误:
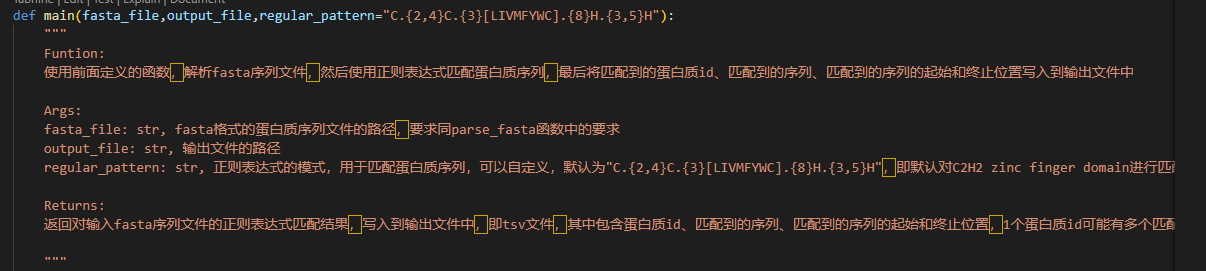
TypeError: first argument must be string or compiled pattern
这表明 regular_pattern 不是一个字符串或编译的正则表达式对象,而是 None 或其他不符合要求的类型。
1,问题原因:
regular_pattern 的默认值未被正确传递:在我的代码中,main 函数的 regular_pattern 参数有一个默认值:
def main(fasta_file, output_file, regular_pattern="C.{2,4}C.{3}[LIVMFYWC].{8}H.{3,5}H"):
如果 args.regular_pattern 是 None(即未通过命令行提供正则表达式参数),main 函数会使用默认值。
但是,如果 args.regular_pattern 被显式传递为 None,则会覆盖默认值,导致 re.finditer 报错。
命令行参数未正确传递:
我在运行脚本时没有提供 --regular_pattern 参数,args.regular_pattern 的值可能是 None。
在这种情况下,main 函数会将 None 传递给 re.finditer,从而导致错误。
2,解决方法:
(1)在 main 函数中处理 None 值:
在 main 函数中,检查 regular_pattern 是否为 None,如果是,则使用默认值:
def main(fasta_file, output_file, regular_pattern=None):
# 其他都不变,只在最开头的时候加入如下几行:
if regular_pattern is None:
regular_pattern = "C.{2,4}C.{3}[LIVMFYWC].{8}H.{3,5}H"
(2)在命令行参数解析时设置默认值
这个是原来的:

修改如下
parser.add_argument("--regular_pattern",
help="正则表达式的模式,用于匹配蛋白质序列,可以自定义,默认为C.{2,4}C.{3}[LIVMFYWC].{8}H.{3,5}H,即默认对C2H2 zinc finger domain进行匹配",
default="C.{2,4}C.{3}[LIVMFYWC].{8}H.{3,5}H")
(3)确保命令行参数正确传递
在运行脚本时,显式提供 --regular_pattern 参数。例如:
python3 prosite_pattern_match.py --fasta_file /lustre/share/class/BIO8402/hw4/ws_215.protein.fa --output_file c2h2_zf_ws_215.tsv --regular_pattern "C.{2,4}C.{3}[LIVMFYWC].{8}H.{3,5}H"

















 1412
1412

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









