




 这篇博客介绍了如何将vcf文件转换为maf格式,适用于肿瘤基因组分析。通过下载并使用vcf2maf工具,结合vep软件,可以方便地进行文件格式转换。文章提供了vcf2maf的使用方法和参数示例,帮助读者理解转换过程。
这篇博客介绍了如何将vcf文件转换为maf格式,适用于肿瘤基因组分析。通过下载并使用vcf2maf工具,结合vep软件,可以方便地进行文件格式转换。文章提供了vcf2maf的使用方法和参数示例,帮助读者理解转换过程。
小胖友,你是否有很多问号,为什么别人都在做群体cohort突变瀑布图了,你还连个输入文件格式都转换不好?

做肿瘤基因组分析的胖友肯定很熟悉vep软件了,体细胞突变检测结果下来跑个vep,竖线分割长列信息cut grep awk split一顿操作猛如虎,最后啥也没搞明白是常态。。想用R包可视化一下,发现输入文件格式需要maf格式,那今天就先讲这个格式转换吧。下次再更画图的方法。
首先打开你高贵高级高雅的谷狗浏览器,下载vcf2maf软件:
下载地址:https://github.com/mskcc/vcf2maf/releases
解压后就可以直接使用vcf2maf.pl这个脚本了,使用的前提是你提前装好了vep哈,这个脚本可以直接用来注释,跑它就不用单独跑vep啦~
然后看一下它的使用方法:
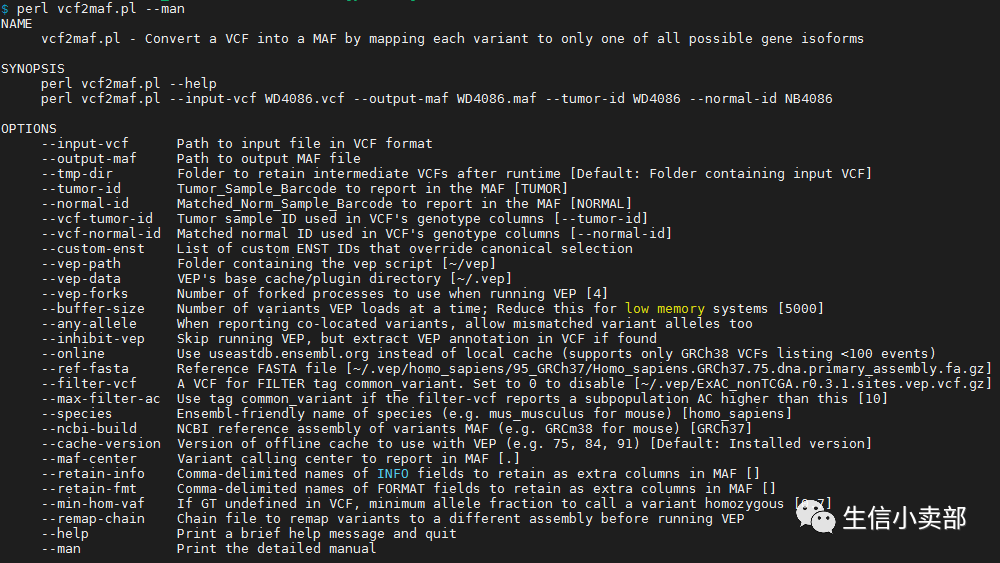
然后照着这个option往里面填参数(这里是示例,真实情况下还是记得写绝对路径哦):
perl vcf2maf.pl \
--input-vcf T668077.oncefiltered.vcf \ #接mutect2跑出来的vcf文件即可
--output-maf T668077.maf \ #需要得到的maf文件名称
--tumor-id T668077 \ #输出的maf文件里面的tumor name
--normal-id N668077 \ #输出的maf文件里面的normal name
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 301
301



 到【灌水乐园】发言
到【灌水乐园】发言







