




 在基因组分析中,针对负链上的基因,文章探讨了先反向互补染色体再截取区间还是先截取区间再反向互补的正确方法。结论是应先截取区间,再进行反向互补操作。通过bedtools工具和Python编程验证,证明了这种方法的准确性。关注更多生信小知识。
在基因组分析中,针对负链上的基因,文章探讨了先反向互补染色体再截取区间还是先截取区间再反向互补的正确方法。结论是应先截取区间,再进行反向互补操作。通过bedtools工具和Python编程验证,证明了这种方法的准确性。关注更多生信小知识。
最近需要根据注释文件在基因组上截取序列,突然想到一个问题:对于下面这样在负链上的基因,我们是先将整条染色体反向互补再截取对应区间?还是先截取对应区间再反向互补呢?

首先亮出答案:先截取区间,再反向互补。比如上面的ALK基因,先截取chr2上的29415640-30144452区间,再反向互补即得到ALK基因的碱基序列。
验证过程:
方法一:bedtools工具包可以根据bed文件提取区间序列,这里以上面的ALK基因为例试一下:
1.首先创建一个bed文件命名为AKL.bed,其中文件第6列可以指定正负链

2.然后运行bedtools,得到ALK.fa序列文件,注意要添加-s选项
bedtools getfasta -fi ucsc.hg19.fasta -bed ALK.bed -s -fo ALK.fa3.比较NCBI上给出的ALK序列和bedtools的运行结果,发现两者一致,说明bedtools工具截取负链上的基因准确,如果不想深究是哪种截取方法,后面直接用这个工具即可
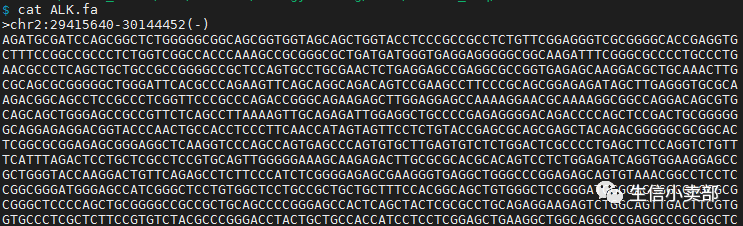

方法二:将两种不
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 6984
6984

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









