




1. 建模:
利用Materials studio2020完成单体的建模
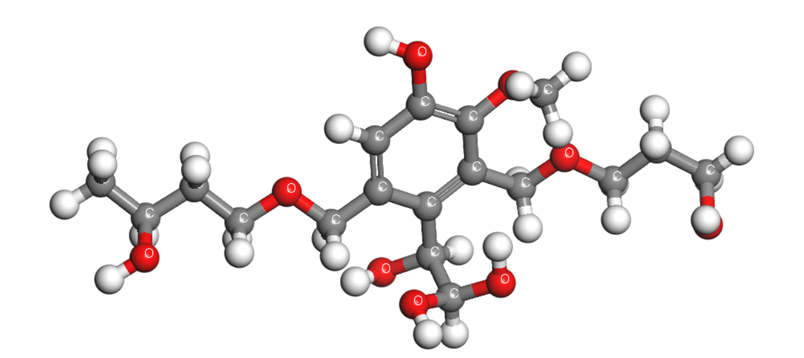
(要注意的是这里的原子没有单独命名,为了后续好生成top,在这里应该将原子名称修改好方便后续写rtp)
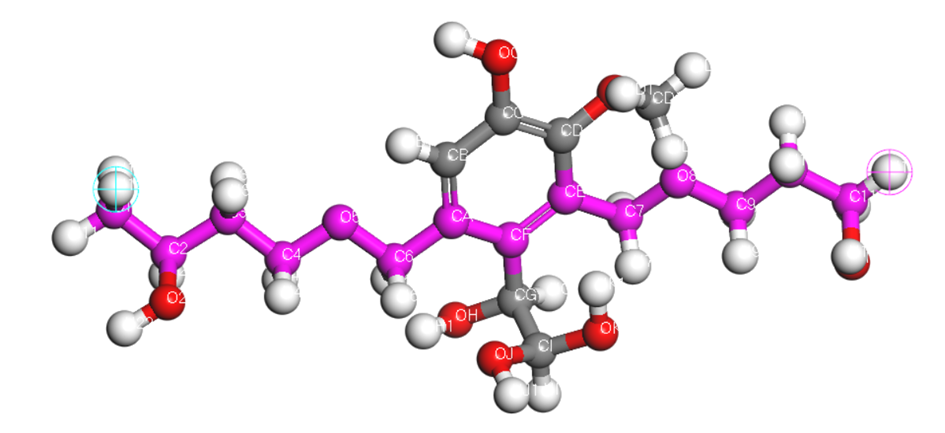
这就是命名好的单体了,蓝色的部分是你选择的repeat unit
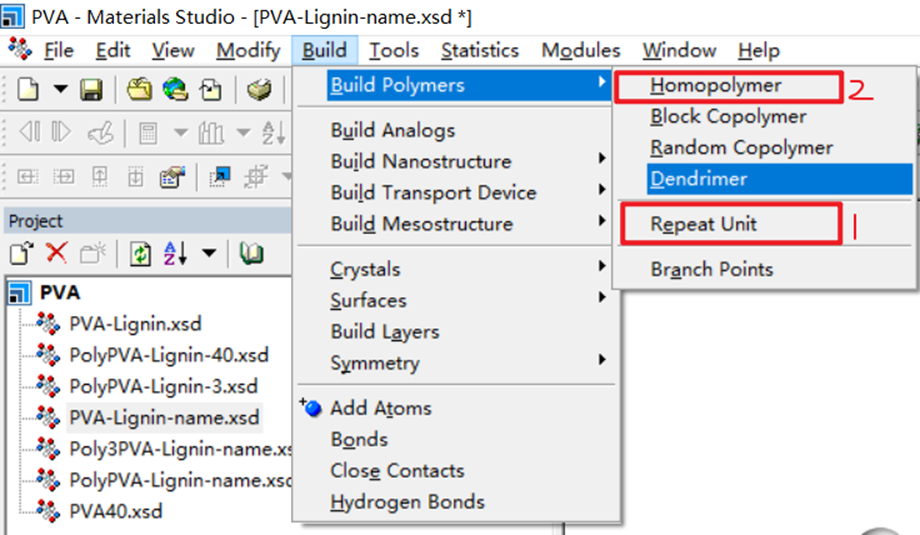
(聚合物的构建方法:可自行搜索)具体怎么构建,这里跳过
通过选择聚合度为40,我们可以构建好命名完整的聚合物大分子,接着我们构建一个聚合度为3的聚合物小分子,以方便我们进行resp的电荷计算,这里的方法我参考的是卢天老师的网页

利用图中的方法,将聚合度为3 的聚合物利用高斯软件进行计算,得到的电荷我们抄录下来(这个电荷我们后续还需要调整)
2. top文件的生成:
首先将聚合度为3的聚合物导出pdb格式,然后打开网站:https://jerkwin.github.io/prog/gmxtop.html
利用这个网站我们可以得到聚合物的原子类型,这里得到的原子类型是opls-aa力场的,我们对应着分子结构和之前命名好的原子名称记录下来(注意:他给的opls-aa的原子类型不完全正确,有些原子会出现错误或者识别不出来,所以最好利用高斯对分子结构进行优化,将优化后的结构放到该网站生成)opls-aa生成的top参数取决于这三个文件:
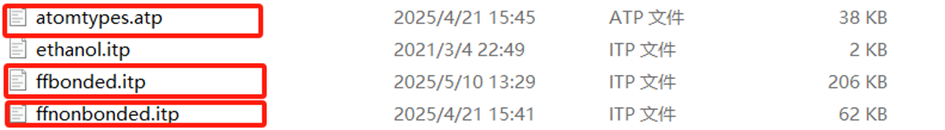
atomtypes.atp:提供原子类型的描述,可以根据分子结构的官能团来寻找对应的原子类型,需要注意的是前65个原子是提供给蛋白质的联合原子
ffbonded.itp:提供原子和原子连接的键长键角二面角信息,需要注意的是他这里使用的原子名称不是opls_xxx,所以我们需要到ffnonbonded.itp里查找opls_xxx对应的原子名称
ffnonbonded.itp:提供原子类型对应的原子名称以及它的sigma和epsilon还有标准电荷的信息等
了解了这个之后,大概就知道gromacs生成的top的键长键角二面角的数据依据,这方便后续了解报错的来源。
开始正式的top生成之路:
书写rtp,首先将头、中、尾定义好,对于我们采用的分子结构,他们的区别就是头部开始是甲基,头部的最后是C11-H11,中部开始不是甲基,中部的最后和头部的最后一样,尾部的开始和中部的开始一样,尾部的最后是C11-H111/H112,明确了这个之后,开始书写rtp,将电荷信息进行修改保证整体为0
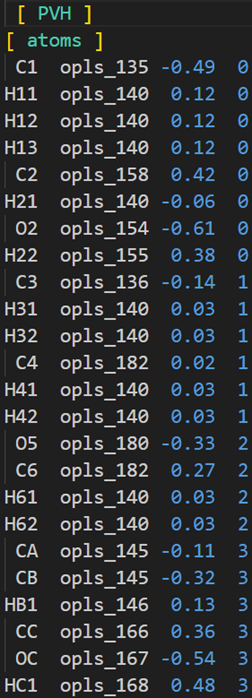
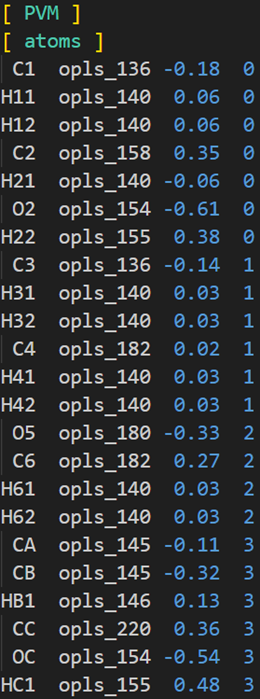
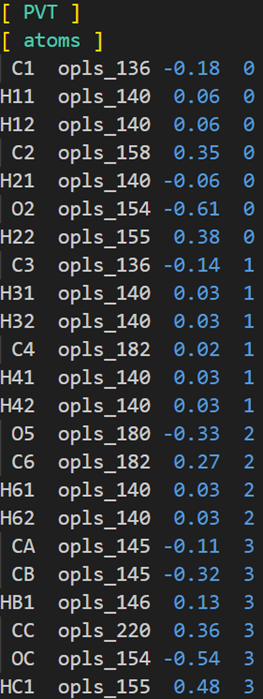
类似这样,将rtp保存到力场文件中
利用gmx pdb2gmx 命令生成top,但这里还要注意:pdb的原子定义!由于Materials studio导出的pdb的原子排序不是按照你书写的rtp的顺序生成的,并且残基名的定义也是他自己定义的,所以这些都需要修改,将同一个残基的原子的残基名改成相同的并将它的残基顺序一并修改,pdb的格式是有要求的,可以自行搜索pdb格式的要求完成pdb的修改,至此我们的基本文件已经修改完成,接着生成出top,这里我们最好还是拿聚合度为3的分子进行测试,因为后面如果出现报错可以方便我们查找对应原子。
这样我们的top就生成好了,接下来就是进行模拟测试top文件的合理性。
3. 测试及报错修改
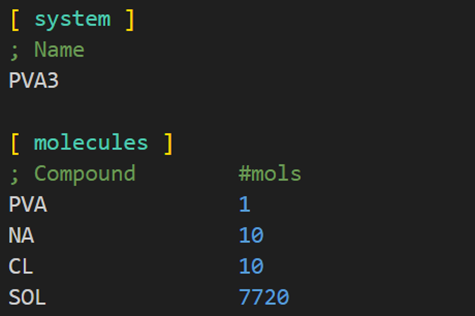
利用PACKMOL将聚合物和水分子装盒子,得到含有水和聚合物的盒子,然后编辑top信息。(类似如下图所示)
进行em,npt,nvt。(需要注意的是,这样产生的top的二面角的funct是1,opls-aa的二面角funt是3,所以我们需要修改二面角信息)
另外如果你的原子类型指认不合理,也就是说力场文件的ffbonded.itp没有你填写的原子类型对应的连接方式的信息,他就会提示你Error:No default xxx,像这种情况,你需要找到它提示的那一行报错,然后抄下来,到ffnonbonded.itp找到该原子对应的原子名称,然后去ffnonbonded.itp找对应的原子名称的连接,比如它提示你对应的原子名称CA-CT-CO的角度没有默认值,你到ffbonded.itp查找CA CT CO也是找不到的,那么此时你需要自行添加该连接方式到里面,至于数据你可以在ffbonded.itp找类似的连接方式,或者利用其他软件生成拓扑将对应的数据填写到上面都是可以的,但是合理行的问题gromacs会自行判断,如果不合理他自己就会中断的,但是我认为如果原子类型指认的足够正确,一般的聚合物分子的连接方式的信息在ffbonded.itp都是可以找到的,也就是说默认的。
回到正题
在测试完聚合度为3的分子之后,保证拓扑的数据不会缺失以及npt、nvt能正常进行后,那么就是将聚合度为40的大分子生成拓扑的问题了。唯一繁琐的地方是pdb的修改,由于Materials studio生成的pdb的原子顺序是乱的,但如果你是聚合物的话,以及按我说的方法生成的聚合物大概率相同残基的原子是在一起的,这就需要你根据pdb最后的CONNECT的信息去自行发现规律了,或者你也可以利用python编写脚本实现自动化排序,但是最后生成的pdb建议不要马上去生成top,而是用可视化软件去查看它的结构是否合理,也许利用脚本生成的pdb压根就显示不出来(我就在这里上过当,不知道是什么原因导致,最后还是乖乖手动排序)
至此你的拓扑就生成完毕了,不过大聚合度的分子往往在某个方向上尺寸特别大,所以需要在水溶液里不断进行em>npt>nvt重复模拟,以形成自然团聚状态,这样也可以减少聚合物尺寸过大带来的盒子过大造成体系分子增加带来计算量过大的问题~~~文中提到的聚合物最开始放在大小为22nm×16nm×12nm的大盒子中,最后自然态下可以放在5nm×5nm×5nm的盒子中。

















 1336
1336

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









