




 Cytoscape是一款开源的网络可视化软件,本文介绍了其安装与基本使用方法,包括安装Java环境,导入并设置节点文件,美化网络图,节点与边的操作,以及网络分析和文件导出。
Cytoscape是一款开源的网络可视化软件,本文介绍了其安装与基本使用方法,包括安装Java环境,导入并设置节点文件,美化网络图,节点与边的操作,以及网络分析和文件导出。
前言
Cytoscape是一款开源软件,可以用于可视化网络和分析。接下来将简单介绍软件的安装与使用。
Cytoscape安装
- 下载软件:Cytoscape
- Cytoscape软件运行需要Java 11 环境支持,在已经版本中需要手动安装,目录版本会自动安装。若软件运行出现问题,可以考虑手动安装Java 11 环境。
- 软件安装无额外注意,可以自行调整安装位置,但要避免出现中文路径。
Cytoscape使用
本文以绘制网络药理学中,中药-成分-靶点网络举例。
- 软件支持csv、xlsx等格式,前期需要准备的文件格式如下:节点文件,note1为化合物、note2为对应的靶点,HGT中药名和对应的化合物,以及对应的属性文件(帮助美化网络)来源可参考:网络药理学

- 点击软件图标运行。先导入节点文件 network.xlsx。选择对应的表,将表中note1设置为绿色圈、note2设置为黄色圈。

- 导入属性文件,选择正确的表,将成分、靶点具体名称设置为note,关键字设置为type。

- 网络图美化。可通过style快速设计节点和线的格式。

然后对具体项进行操作。这里对节点的形状进行操作,根据导入的属性文件,对所对应关键字的节点进行统一操作。

节点的颜色
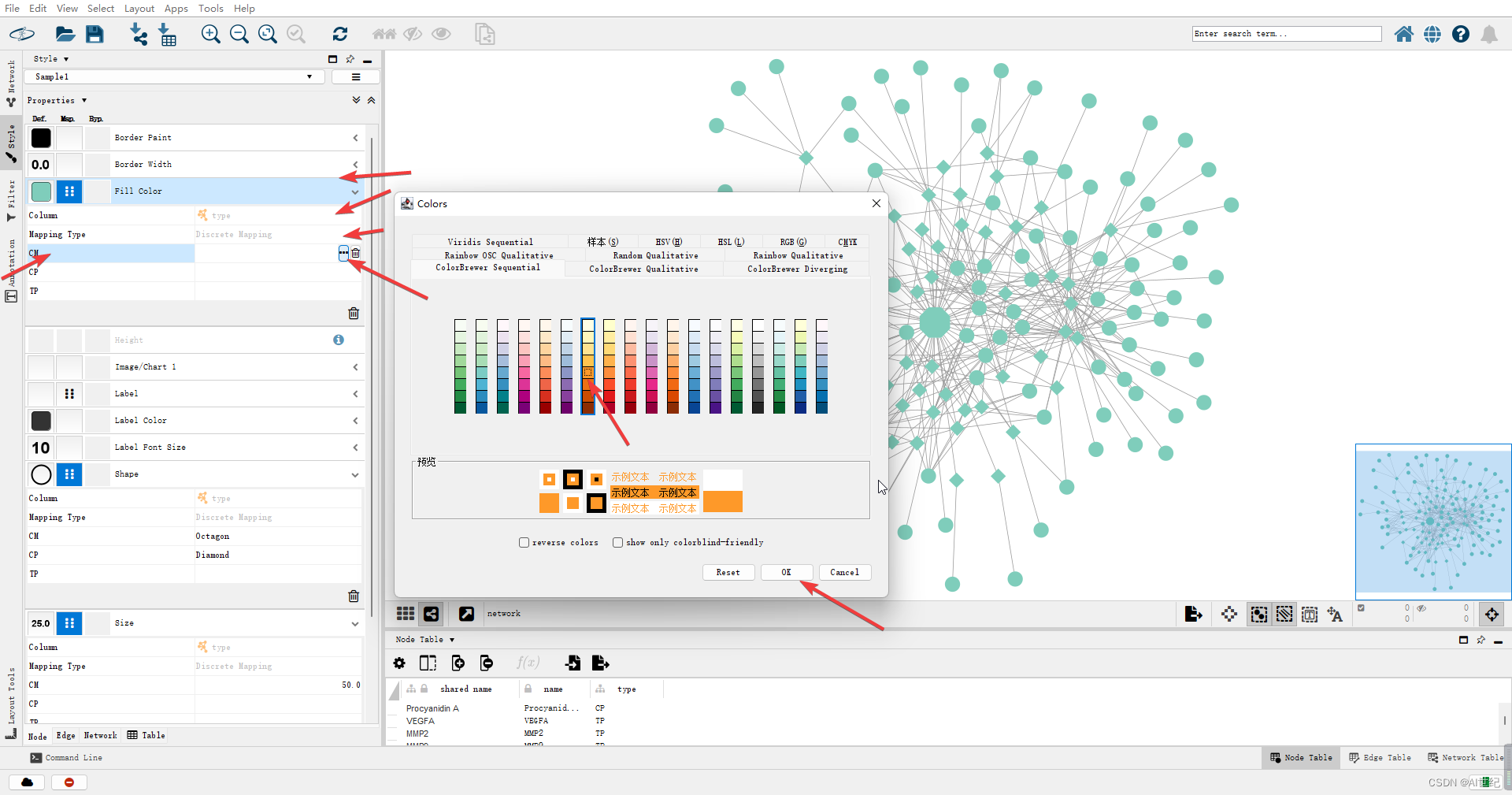
通过上述的步骤,可对其余项进行操作。
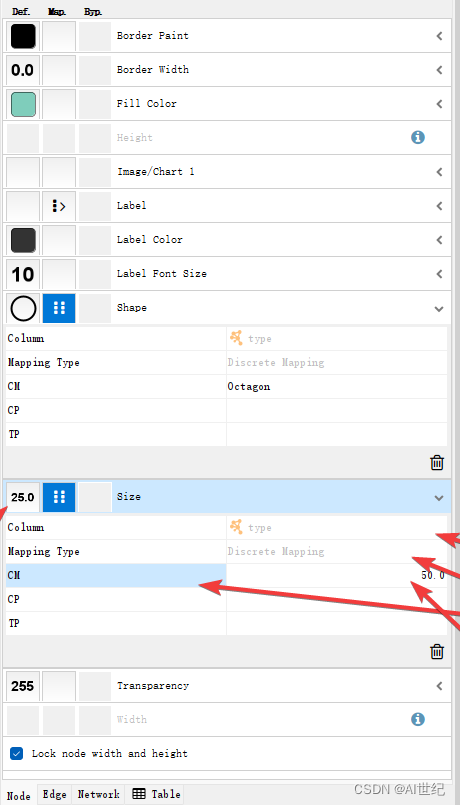
此外,也可以选择edge,对节点这间的线进行操作。
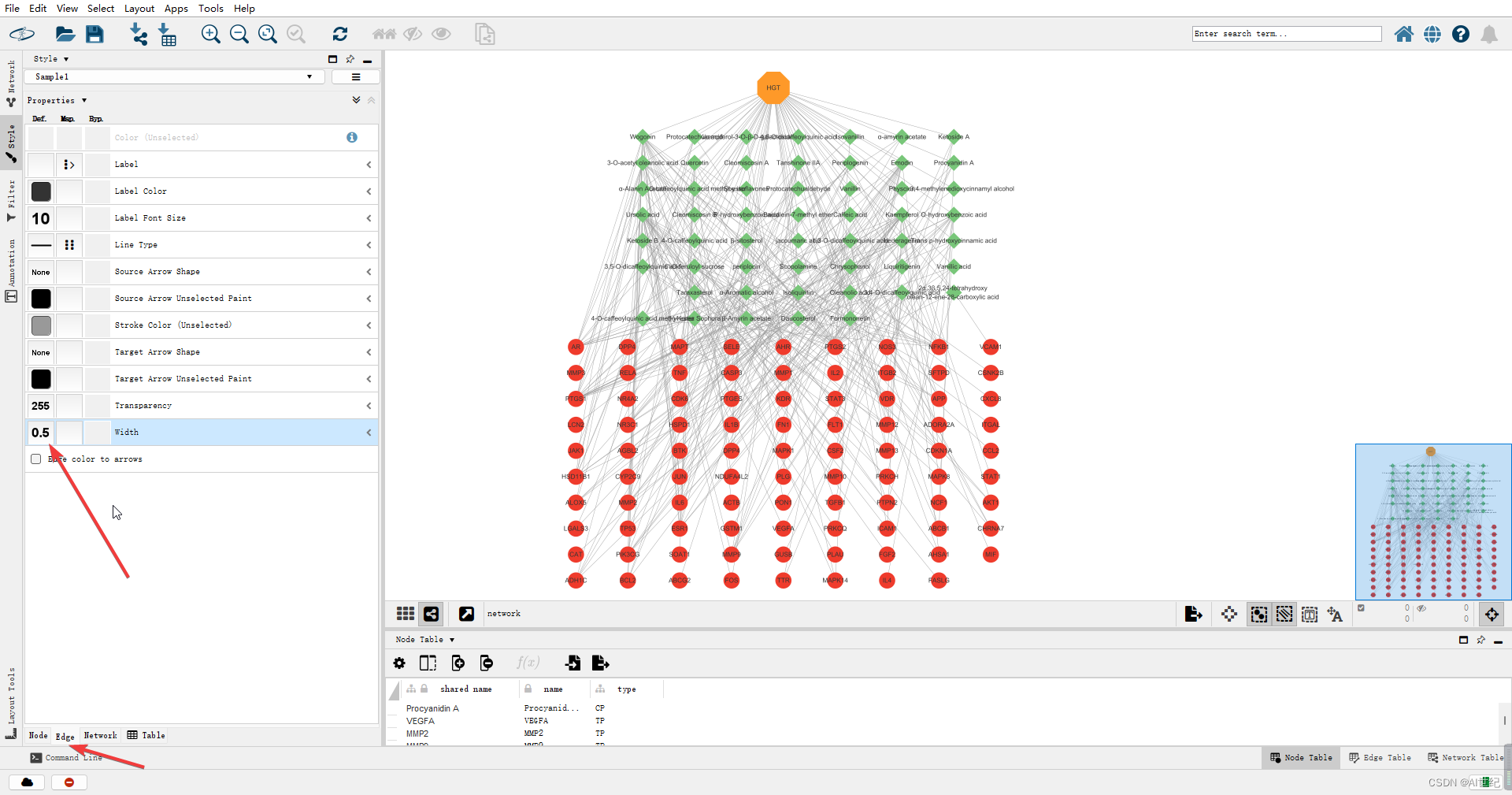
对节点进行排列。

可以对选择项进行节点排序。

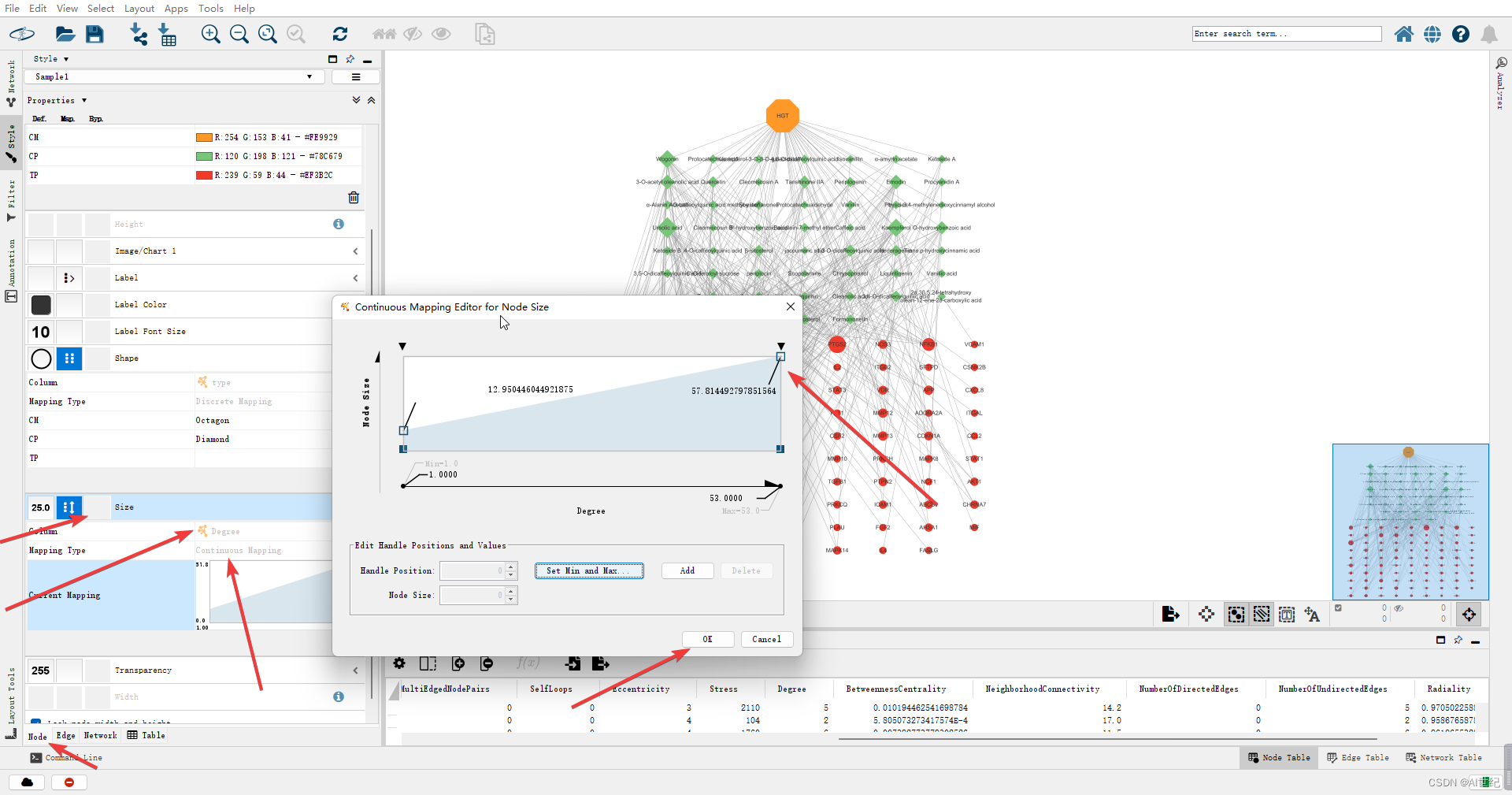
- 根据节点之间的连接degree,绘制节点的大小。首先对网络进行分析。

当再次进行节点大小设计时,会有degree,根据它可调整节点的大小。
- 文件导出。这里建议保存好工作文件.cys。可以也出网络分析的数据,导出网络可视化的图。

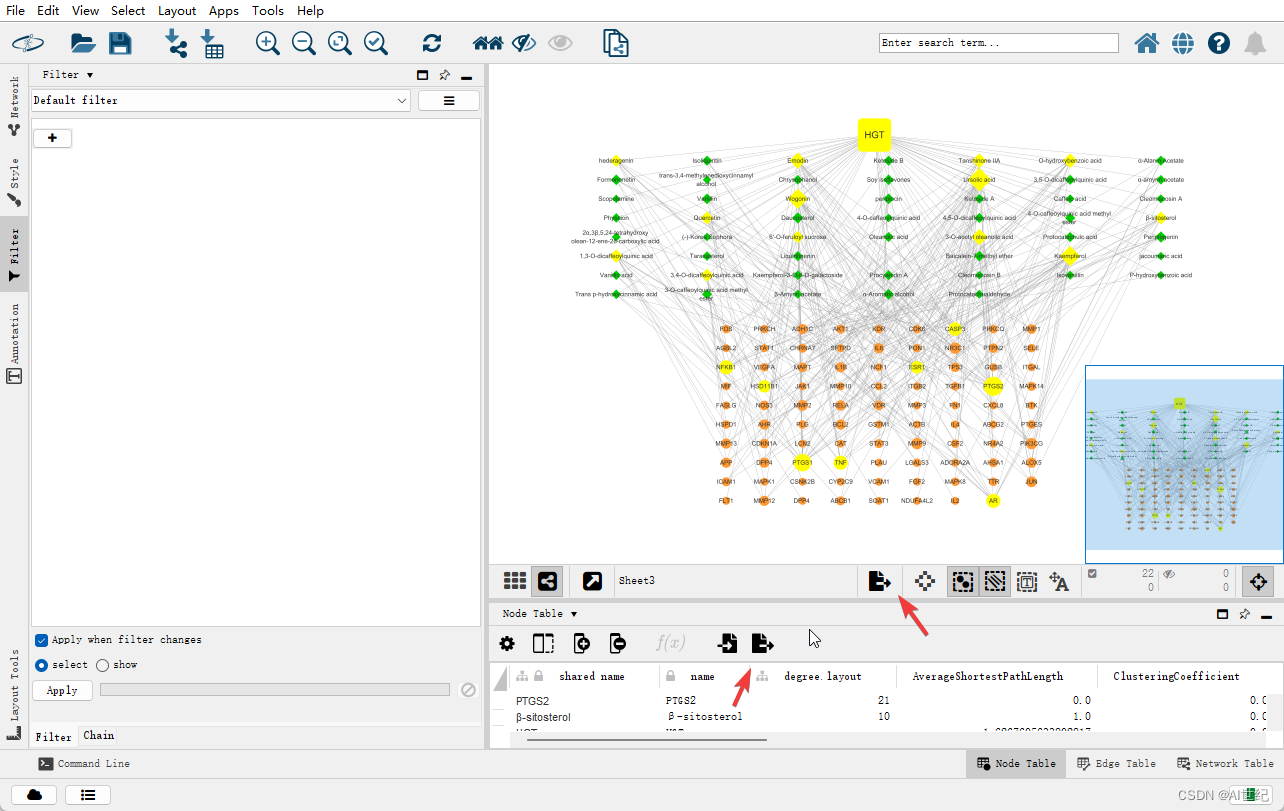
导出为png格式。

以上为Cytoscape软件的简单操作,更多的使用可以参考:官方教程

















 2543
2543

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









