




 AMDock结合Autodock工具,支持多种功能,尤其适合处理含锌离子的蛋白质。对接流程包括准备受体和配体结构,选择合适的对接空间,最后通过PymOL进行结果可视化。提供详细的操作指南,从UniProt获取蛋白质结构,ZINC下载配体,到使用AMDock进行分子对接。
AMDock结合Autodock工具,支持多种功能,尤其适合处理含锌离子的蛋白质。对接流程包括准备受体和配体结构,选择合适的对接空间,最后通过PymOL进行结果可视化。提供详细的操作指南,从UniProt获取蛋白质结构,ZINC下载配体,到使用AMDock进行分子对接。
分子对接
AMDock集成了Autodock Vina 和 Autodock4、ADT scripts、AutoLigand、Open Babel、PDB2PQR 和 PyMOL 的功能。对于活性位点中含有锌离子的蛋白质,AMDock可以选择使用专门定制的 Autodock4Zn参数。
- 对接流程图

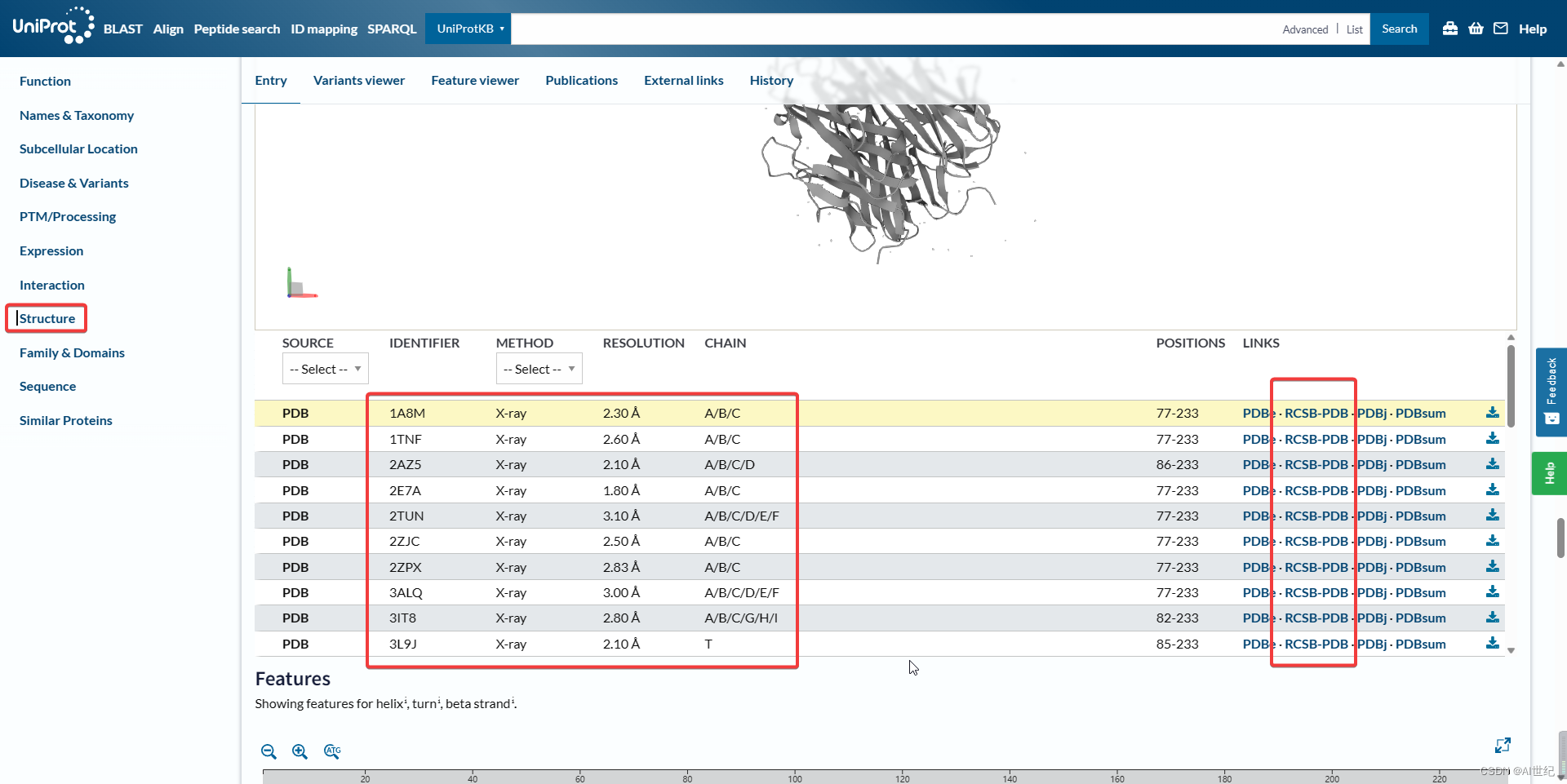
- 首先需要准备分子对接的受体结构。使用 UniPort 数据库可以查询蛋白质的相关信息。在检索框检索蛋白质,在 Structure 查看蛋白质结构信息。

结构信息包含 X-ray、HMR 解析的结构,点击用RSCB-PDB 可以在PDB数据库下载蛋白质结构。
关于分子对接中受体的选择:
- 根据分子对接的用途选择是否为人源蛋白质。
- 蛋白质解析的结构是未突变。
- 蛋白质分辨率越小越好。
- 蛋白质结构需要完整,有一些结构只解析其中的某一个亚基。
- 有配体参与的蛋白质结构。
- 在PDB数据库中,可以查询到发表蛋白质结构的文献。


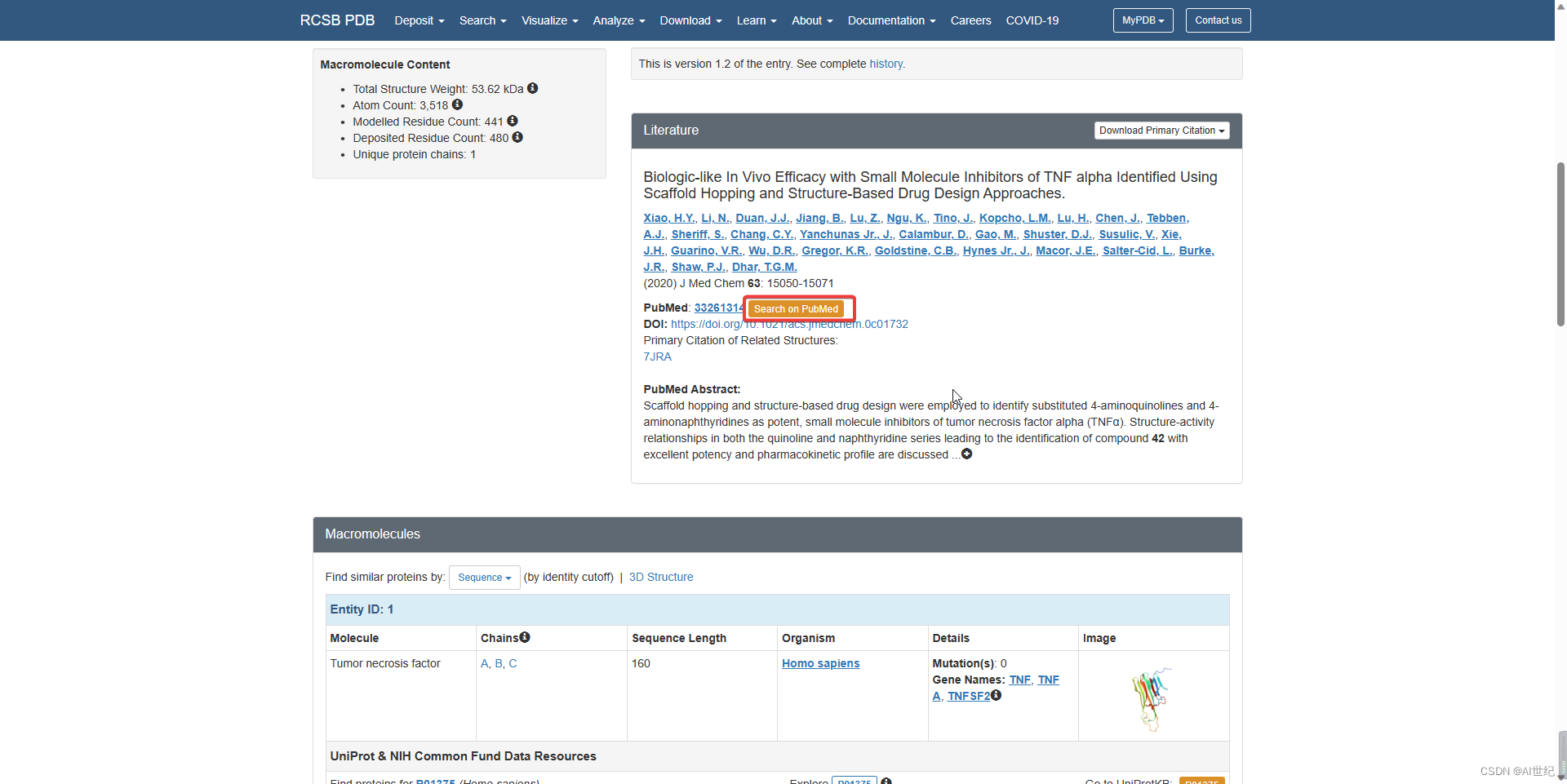
下载蛋白质结构,文件格式为 pdb。

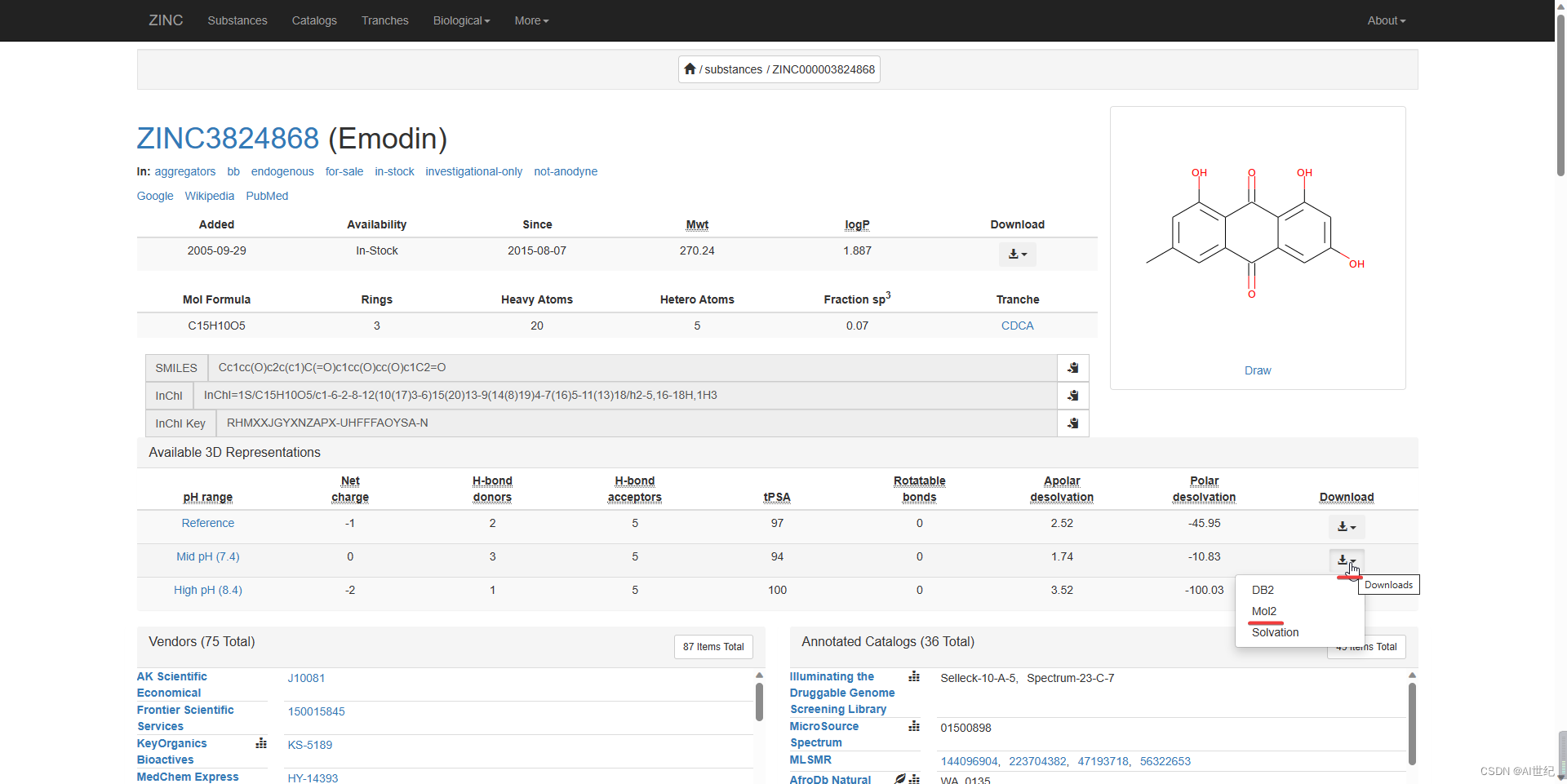
- 准备配体结构,使用 ZINC 数据库下载配体的结构,格式为 mol2。如果配体结构无法在 ZINC 数据库中查询,可以通过Chem 3D 软件进行绘制,保存为mol2格式。

- 使用AMDock进行分子对接。AMDock软件解压后就可以使用。
- 序号1、2、3创建分子对接的文件目录
- 序号4、5、6将受体和配体导入软件,并进行预处理。
- 序号7、8是选择受体的对接空间,第一个是软件自动预测蛋白质的对接空间,需要花费较长时间。第二个是根据文献查询蛋白质的那些氨基酸是对接位点,软件这些氨基酸来确认对接空间。第三个是根据蛋白质中已存在的配体确认对接空间(蛋白质的已存存配体在分子对接时是去掉的,不会影响新配体的对接。)。第四个是自己手动输入,自定义对接空间。
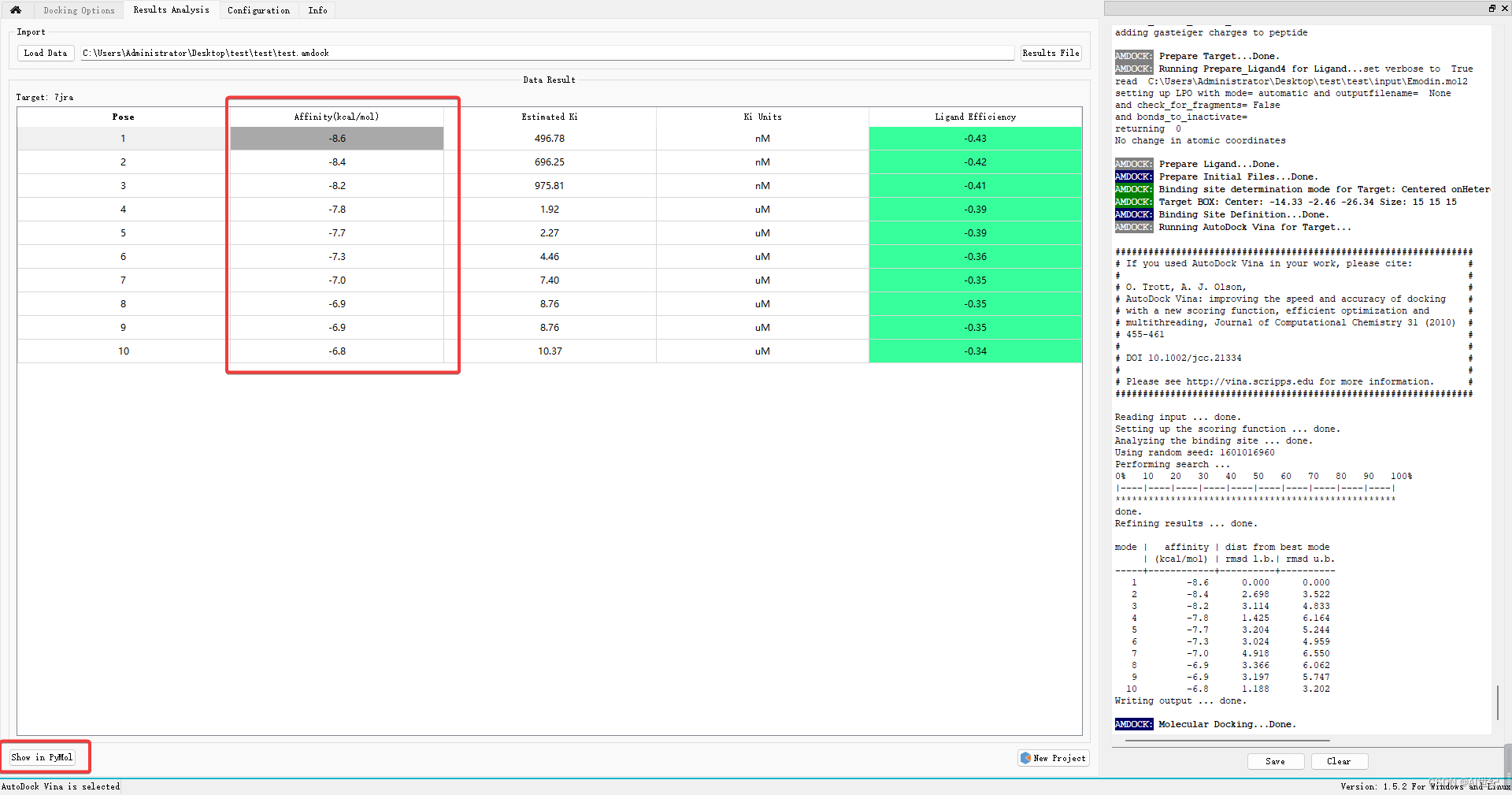
- 当上述处理无问题时,序号9进行分子对接。


- 分子对接完毕后,可以看到分子对接的结构,一般选择第一个结果。点击show Pymol 可以可视化对接结果,并可以进行分子对接可视化处理。如何使用pymol进行可视化,参考PyMOL作图
| 数据库 | 网址 |
|---|---|
| UniPort | https://www.uniprot.org/ |
| PDB | https://www.rcsb.org/ |
| ZINC | https://zinc15.docking.org/ |
以上是AMDock进行分子对接的简单操作。

















 3357
3357




 到【灌水乐园】发言
到【灌水乐园】发言







