



宝子们!最近医学圈被一组“双疾病CP”刷屏了!心血管疾病(CVD)和抑郁症这对“难兄难弟”在顶级期刊《Molecular Psychiatry》上演了一出“虐心大戏”,原来心脏的痛和心灵的伤竟能互相“传染”?😱
传统研究总爱“单打独斗”,但西南医科大学团队这次玩了个大的!他们用“Meta分析+孟德尔随机化”双剑合璧,一口气分析了39项研究、6万+患者数据,还搬出基因层面的因果论证,直接把双疾病关联研究送上热搜。
Meta+MR的"黄金组合"已助300+学者斩获高分论文!✔️进阶玩法:叠加"代谢组+蛋白组"双引擎,再注入机器学习算法,这种"科研超频"模式让90%用户实现影响因子突破5+。
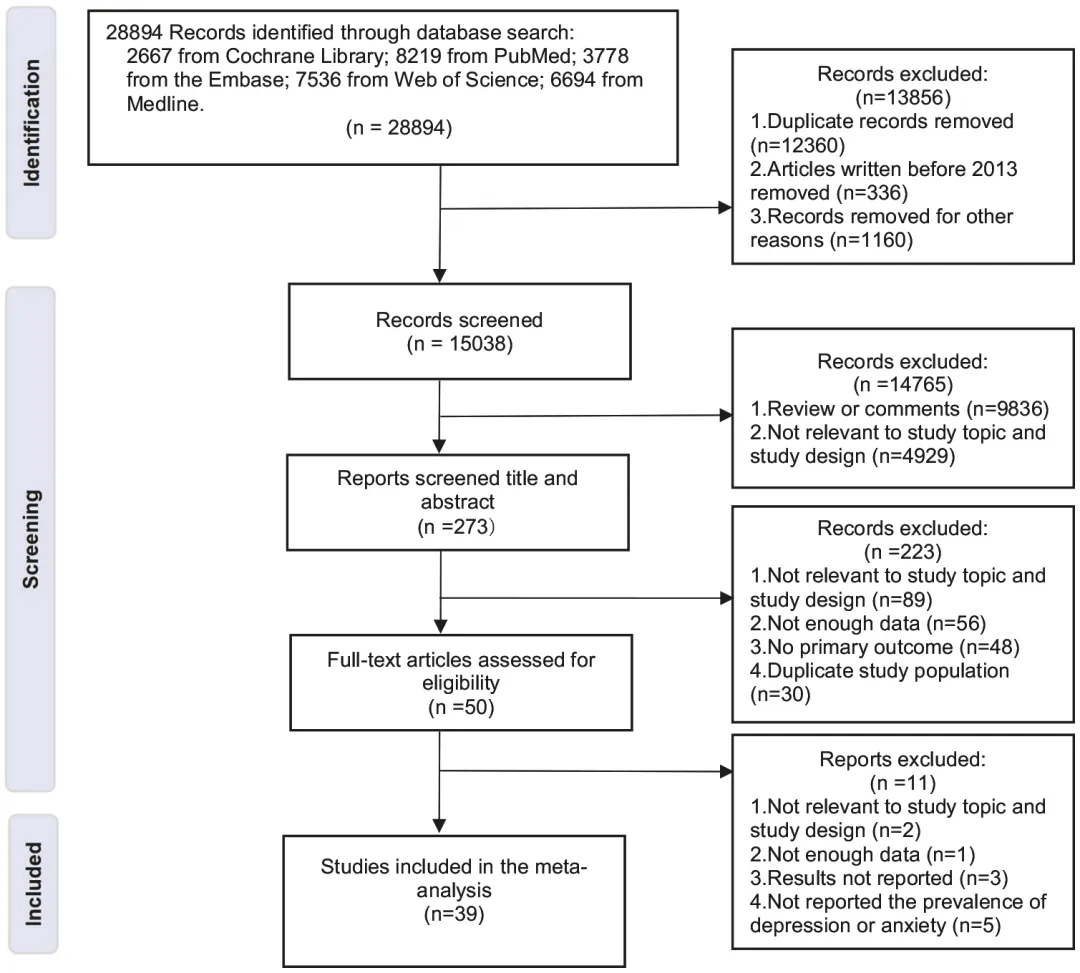
让我们一起解析这篇由西南医科大学团队发表在中科院一区的高质量论文。Meta分析揭示了心血管疾病(CVD)与抑郁症之间的相关性,而MR分析进一步探索了它们之间是否存在因果关系。两种方法相互补充,为研究结论提供了更扎实的证据支持。别看整篇文章只展示了三张图,却已经充分体现了联合分析的强大力量!这类研究套路值得大家收藏参考!

题目:心血管疾病与抑郁症:一项Meta分析与孟德尔随机化分析研究
杂志:MOLECULAR PSYCHIATRY
影响因子:IF=9.6
中科院分区:医学一区
发表时间:2025年4月
01 研究背景
心血管疾病(CVD)患者中抑郁症的发病率显著高于普通人群,这种共病关系已被多项研究关注。抑郁不仅影响患者的生活质量,还可能增加心肌梗死、中风及死亡的风险。然而,CVD与抑郁之间是否存在因果关系,尚缺乏明确结论。本研究通过整合Meta分析和孟德尔随机化(MR)分析两种方法,从相关性到因果性层层推进,进一步探讨抑郁对CVD的潜在影响。同时,研究还利用多种大规模GWAS数据库,为理解其生物学机制和临床干预策略提供了坚实依据。
02 研究思路
本研究通过Meta分析整合了39项观察性研究,评估CVD患者中抑郁和焦虑的患病率及其对全因死亡率的影响,采用随机效应模型处理异质性,并验证结果稳健性。
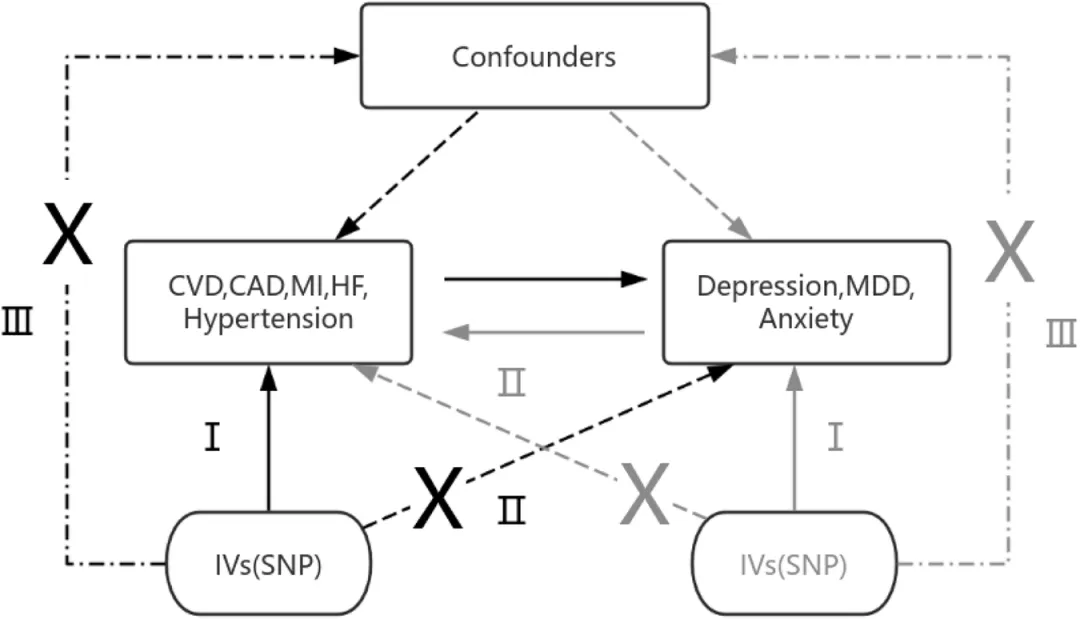
为探讨CVD是否与抑郁或焦虑存在因果关系,研究进一步采用基于公开GWAS数据的双样本双向孟德尔随机化(MR)分析,严格筛选满足三大假设的SNP作为工具变量,并统一暴露与结局的等位基因信息。
主要分析方法为反向方差加权(IVW),同时结合加权中位数法和MR-Egger回归。使用MR-PRESSO方法检测并剔除多效性SNP,确保分析结果不受偏倚。Cochrane's Q检验和MR-Egger截距用于评估异质性和水平多重效应。
为控制多重检验误差,采用FDR校正,FDR<0.05为显著结果,未通过FDR但P<0.05的结果为提示性关联。还通过逐一排除分析和散点图对结果进行稳健性验证和可视化。
所有分析均使用R 4.3.2版本完成,主要依赖“TwoSampleMR”和“MR-PRESSO”包。
03 研究结果
1.心血管疾病患者抑郁症、焦虑症患病率及全因死亡率的荟萃分析
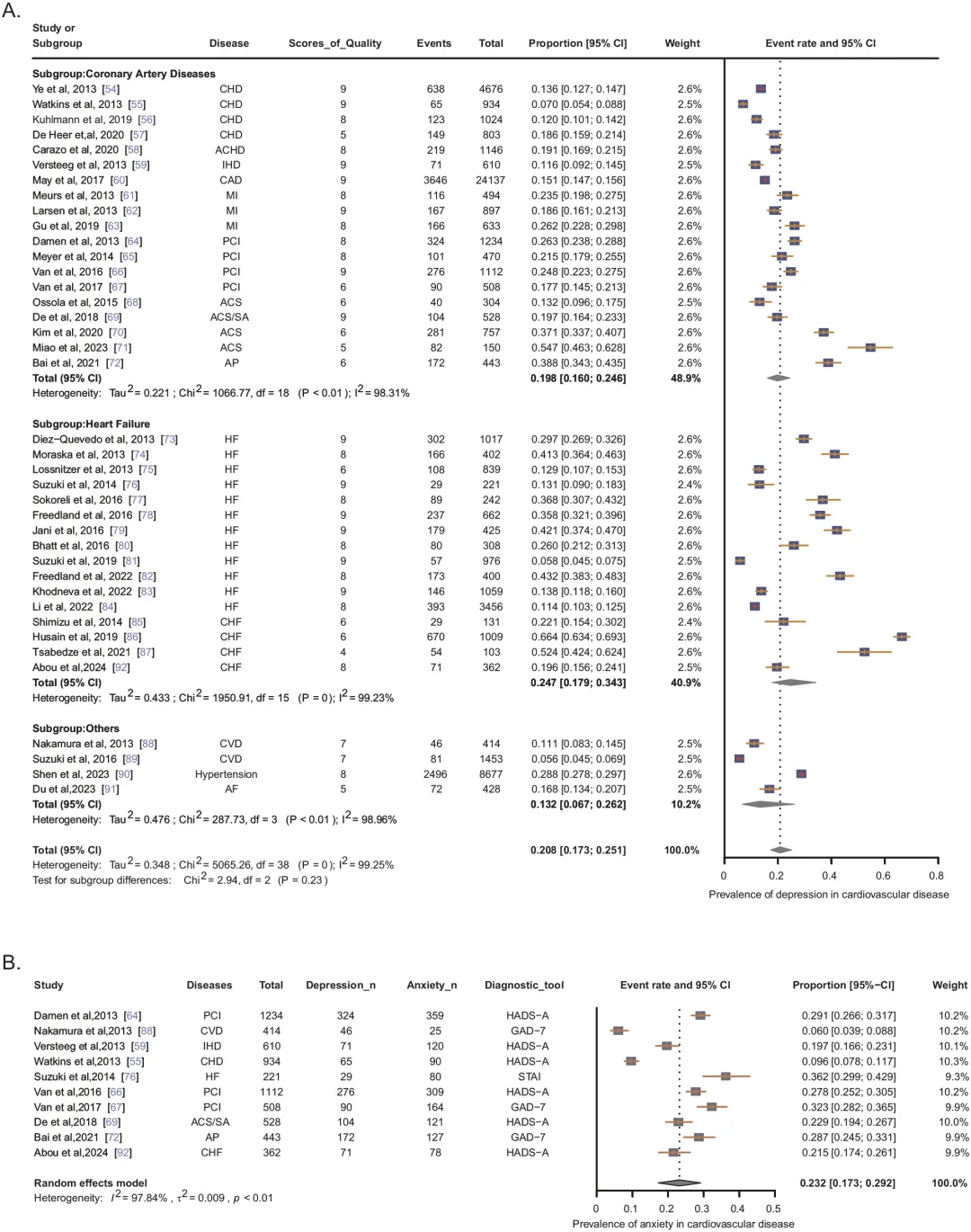
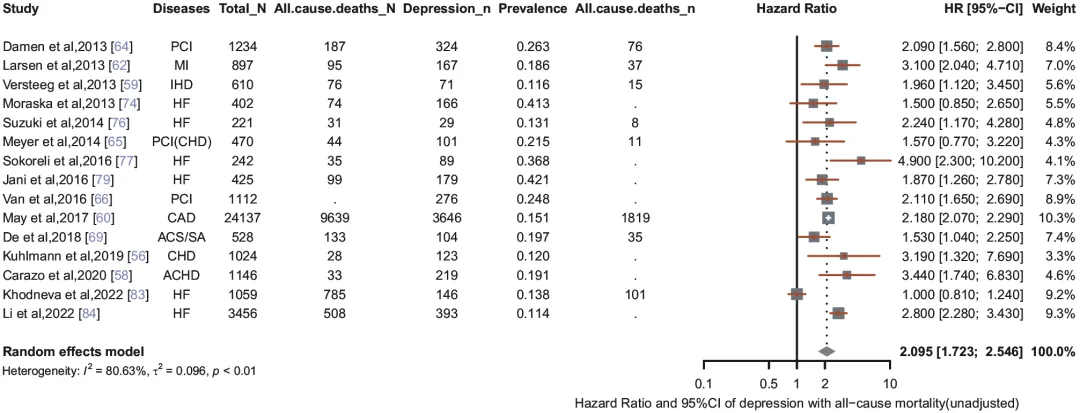
本研究纳入了39项研究,评估了心血管疾病(CVD)患者中抑郁症和焦虑症的患病率及其与全因死亡率的关系。结果显示,CVD患者中抑郁症的总体患病率为20.8%,其中冠状动脉疾病(CAD)患者为19.8%,心力衰竭患者为24.7%;CVD患者中焦虑症的患病率为23.2%。抑郁症状与全因死亡率显著相关,未调整风险比为2.095。考虑到研究间存在高度异质性(I²=99.25%),研究还进行了一系列亚组分析,按疾病类型、大洲分布及抑郁评估量表等因素进行了深入探讨,确保结果的稳健性。
2.孟德尔随机化结果
通过MR-PRESSO去除异常值后,整体CVD与抑郁症及焦虑症之间未发现显著关联。然而,亚型分析揭示,冠状动脉疾病、心肌梗死、心力衰竭和高血压与抑郁症有显著关联,其中仅高血压的关联通过FDR校正。反向MR分析进一步表明,抑郁症显著增加多种CVD的风险,且经多重检验校正后依然显著。相反,焦虑症仅与心力衰竭存在微弱关联,校正后不再显著。敏感性分析显示无水平多效性(P>0.05),且F统计量大于10,表明工具变量强度足够,结果稳健。这些发现表明,抑郁症可能是CVD的独立危险因素,而焦虑症与CVD的因果关系较弱。

04 文章总结
通过meta分析和MR分析,作者揭示了抑郁症与心血管疾病(CVD)之间的双向因果关系。研究表明,抑郁症显著增加冠心病、心肌梗死和心力衰竭的风险,并强调了抑郁症管理在改善心血管预后方面的重要性。
05 小优结语
Meta分析×孟德尔随机化=学术盲盒开挂指南!首先,Meta分析能通过整合多个研究数据,提高统计效力,扩大样本量,让结果更有说服力。而MR通过遗传变异来推断因果关系,减少了混杂因素的影响,得出的结论更加精准!两者结合,能同时验证关联和因果关系,给你双重保险,让结论更加实锤!

















 1342
1342

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









