



机器学习算法+多组学
孟德尔随机化(MR)如今的热度堪称席卷生信领域,俨然成为新的“顶流”。
不得不感叹,这项技术真的有点“后来者居上”的味道!早期生信领域的核心是基因筛选,但现在MR分析不仅强势入局,还直接成为了这块地盘的新主力。✨
所以,别再用老观念看待MR分析了。与其说它抢占了生信阵地,不如说它是为研究提供了全新的突破口。你完全可以把它当作一件“科研利器”——和单细胞分析、机器学习一样,MR分析能轻松融入各种研究思路,拓展研究方向的广度和深度。
接下来给大家分享一篇用孟德尔随机化(MR)筛选基因的创新研究案例:这篇文章由首都医科大学团队发表,独辟蹊径地利用MR分析筛选出乳腺癌中与免疫细胞相关的关键基因。随后,他们结合转录组数据进行验证,进一步强化了研究结果的可信度和科学性。

题目:乳腺癌中免疫细胞相关基因的挖掘:基于汇总数据的孟德尔随机化分析与共定位研究
杂志:Breast Cancer Res
影响因子:IF=6.1
中科院分区:医学一区
发表时间:2024年11月
01 研究背景
乳腺癌是全球女性中最常见的癌症类型,包含多种亚型,不同亚型需要采用不同的治疗策略。肿瘤微环境和免疫反应在乳腺癌的发生和进展中具有重要作用。然而,关于乳腺癌特异性免疫细胞内的基因相关研究仍然较少,证据有限。
02 方法学
本研究利用Yazar等的eQTL数据及BCAC的GWAS数据,探索特定免疫细胞中基因表达与乳腺癌的潜在因果关联,并用FinnGen的GWAS数据验证结果。此外,通过bulkRNA-seq数据库进行差异基因表达分析,识别乳腺癌组织中的差异表达基因。
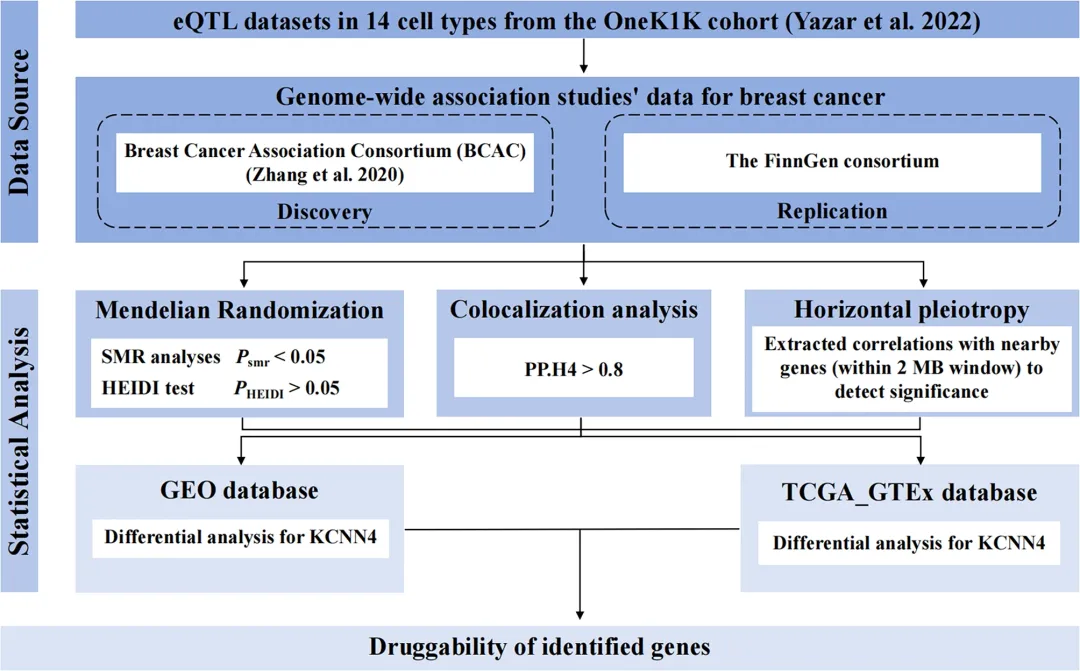
暴露数据来源
本研究使用的eQTL数据来源于OneK1K队列,具体数据来自Yazar等人的研究。该队列包括982名供体的1.27百万外周血单个核细胞(PBMCs),并使用单细胞RNA测序(scRNA-seq)数据进行分析。研究从14种不同的免疫细胞类型中鉴定出26,597个独立的顺式eQTL,包括CD4 + 初始和中心记忆T细胞(CD4NC)、具有效应记忆或中心记忆表型的CD4 + T细胞(CD4ET)、表达SOX4的CD4 + T细胞(CD4SOX4)、具有效应记忆表型的CD8 + T细胞(CD8ET)、未成熟和中心记忆的CD8 + T细胞(CD8NC)、表达S100B的CD8 + T细胞(CD8S100B)、自然杀伤细胞(NK)、募集型NK细胞(NKR)、浆细胞、记忆B细胞(Bmem)、未成熟和初始B细胞(Bin)、经典单核细胞(MONOc)、非经典单核细胞(MONOnc)以及树突状细胞(DC)。
结局数据来源
乳腺癌GWAS汇总数据来自乳腺癌协会联盟(BCAC),包括133,384例病例和113,789名对照,以及18,908名携带BRCA1突变的欧洲血统个体。BCAC不仅研究整体乳腺癌风险,还探讨了亚型特征,包括Luminal A、Luminal B、Luminal B HER2Neg、HER2 Enriched和三阴性(Triple Negative)亚型。为提高对三阴性亚型的敏感性,Zhang等进行了BCAC和CIMBA BRCA1突变携带者的荟萃分析(BRCA-TN亚型)。
验证分析使用了FinnGen联盟的GWAS数据,涵盖18,786名乳腺癌患者和182,927名对照,来自芬兰生物样本库的超过50万份样本。
bulk RNA测序数据来源
从基因表达综合数据库(GEO)获取了微阵列测序数据GSE162228。从UCSC Xena中下载了来自癌症基因组图谱(TCGA)的乳腺癌数据集,包含1,104个乳腺肿瘤样本和113个正常乳腺组织样本。此外,还使用了基因型-组织表达(GTEx)数据库,包含179个正常乳腺组织样本。
遗传工具选择
在SMR分析中,使用cis-eQTL遗传变异作为工具变量(IV)来评估基因表达。
统计分析
基于汇总数据的孟德尔随机化与工具变量依赖性异质性检验。
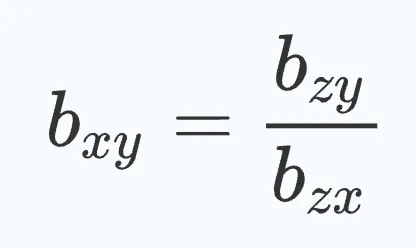
共定位分析
为了揭示连锁不平衡的混杂效应 [16],并检验基因与乳腺癌之间是否存在共享的因果变异,我们使用“coloc” R包进行共定位分析。分析设定了五种假设:
① 零假设(H0):基因或乳腺癌均不存在因果变异;
② 假设1(H1):仅基因存在单一因果变异;
③ 假设2(H2):仅乳腺癌存在单一因果变异;
④ 假设3(H3):基因和乳腺癌各自存在两个独立的因果变异;
⑤ 假设4(H4):基因和乳腺癌共享一个因果变异。
所有显著基因(PSMR < 0.05,PHEIDI > 0.05)来自不同细胞类型并进行共定位。分析采用默认的先验概率(PP.H1 = 1 × 10⁻⁴,PP.H2 = 1 × 10⁻⁴,PP.H3 = 1 × 10⁻⁵)。如果H4假设的后验概率超过80%,则表明基因与乳腺癌之间高度可能存在共定位关系。
评估水平多效性
为消除水平多效性,检查工具变量与邻近基因的相关性(2MB窗口内),若P < 5 × 10⁻⁸则进行SMR分析,数据不足时使用GWAS目录。未满足条件的基因无法排除水平多效性。
基于GEO和TCGA_GTEx数据库的差异基因鉴定
使用“limma”方法识别GSE162228中肿瘤和正常样本的差异基因,筛选标准为P < 0.05且|log2 FC| > 0.5。结合GTEx和TCGA数据补充正常样本,修正批次效应后,使用Wilcoxon检验,P < 0.05为显著,绘制箱线图。
可药物化基因鉴定
在DrugBank、Dependency Map和ChEMBL数据库中查找乳腺癌相关的靶向药物,并通过PubMed等数据库进行进一步检索。
数据分析使用R v4.2.3和SMR v1.3.1。
03 结果
SMR和HEIDI测试
在FDRSMR < 0.05和PHEIDI > 0.05的阈值下,团队识别出116个与乳腺癌及其亚型相关的基因,涉及14种细胞类型,主要集中在CD4nc、NKr、CD8et、CD8nc和CD4et细胞中。
与整体乳腺癌相关的前三个基因为:CD4nc中的L3MBTL3(PFDR = 2.11 × 10⁻⁶)、CD4nc中的MRPS18C(PFDR = 3.19 × 10⁻⁶)和MONOnc中的KCNN4(PFDR = 3.19 × 10⁻⁶)。对于Luminal A型乳腺癌,前三个基因为KCNN4(PFDR = 4.04 × 10⁻⁵)、TNNT3(PFDR = 3.73 × 10⁻⁴)和SCAMP3(PFDR = 4.48 × 10⁻⁴)。对于三阴性乳腺癌,前三个基因为MDM4(PFDR = 2.50 × 10⁻⁷)和Bin(PFDR = 5.78 × 10⁻⁷)、RPS18(PFDR = 6.07 × 10⁻⁷)。BRCA-TN型乳腺癌的前三个基因为RPS18(PFDR = 6.59 × 10⁻⁹)、CD8nc(PFDR = 6.59 × 10⁻⁹)和MDM4(PFDR = 6.59 × 10⁻⁹)。其他亚型未发现显著基因。
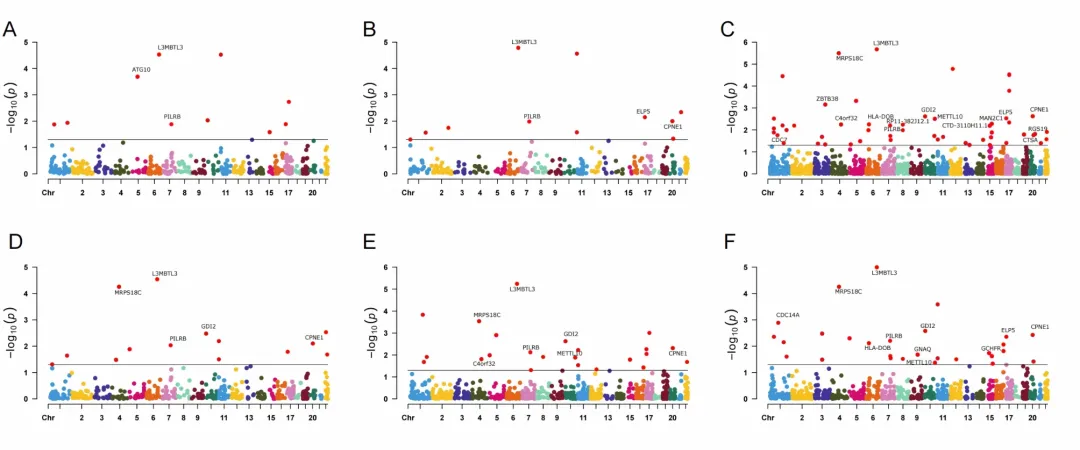
复制分析
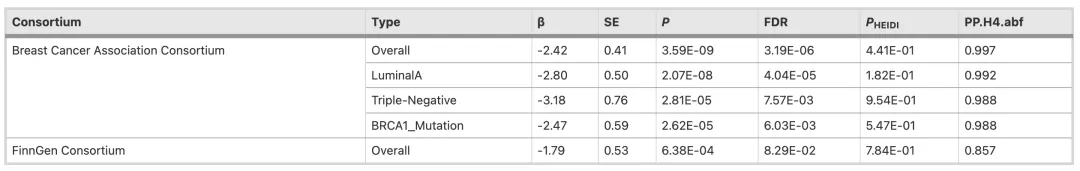
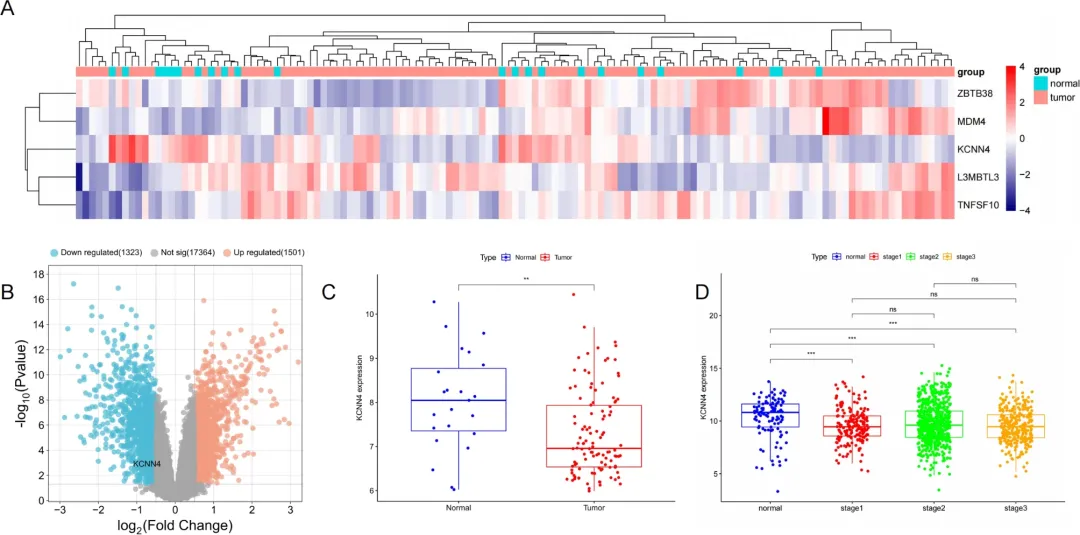
使用芬兰基因组联盟(FinnGen)和免疫细胞eQTL数据进行分析,筛选出73个基因。共定位分析发现13个基因与乳腺癌潜在相关,其中CNN2、FIBP和KCNN4基因表达与乳腺癌相关,KCNN4在BCAC和FinnGen数据库中均显示潜在因果关系。
乳腺癌相关差异基因的鉴定
筛选了GSE162228的基因测序数据,鉴定出2824个与乳腺癌相关的差异表达基因(DEGs),其中包括1501个上调基因和1323个下调基因。团队发现KCNN4基因在肿瘤和正常组织之间表现出差异表达,这与SMR分析结果一致(log2FC = -0.735,P = 0.001)。乳腺癌样本中KCNN4的相对表达水平显著低于正常样本(P < 0.05)。这一结果与GEO数据库中的差异基因分析结果一致。
04 讨论
总的来说,该团队成功识别了17个与乳腺癌及其亚型相关的基因,涉及9种不同的免疫细胞类型。这些基因表现出不同的效应大小,并与乳腺癌亚型有不同的关联,提供了免疫细胞特异性基因在癌症发展中的潜在作用的见解,为未来的靶向干预或治疗铺平了道路。同时,这些基因的识别为基因靶向药物的开发开辟了新途径。通过精确靶向这些基因,有可能设计出更有效且毒性较小的治疗药物,这可能改善现有的乳腺癌治疗方法,并为患者提供更多的生存和生活质量改善的选择。
05 小优结语
结合SMR、HEIDI和共定位分析,深入探讨免疫细胞特异性基因与乳腺癌的因果关系,并通过GEO、TCGA和GTEx等数据库增强研究可靠性。它为基因靶向药物开发提供了潜在靶点,有助于设计更精准的治疗方案,同时通过跨数据库验证提高了结果稳健性,为乳腺癌精准医学和药物研发提供了有力支持。

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









