




题目:SETD2缺失扰乱肾癌的表观遗传格局,促进转移,并产生对组蛋白伴侣复合物的可操作依赖性
期刊:Nature Cancer
发表时间:2022.2
影响因子:23.177
技术手段:ATAC-seq,RNA-seq,ChIP-seq,ChIP-qPCR,RT-qPCR等技术
DOI号:doi.org/10.1038/s43018-021-00316-3
一、研究背景
SETD2是一种RNA聚合酶II(Pol II)相关的组蛋白甲基转移酶,催化H3K36me2的甲基化,从而在转录活跃区域产生H3K36me3修饰。SETD2是不同癌症类型中最常突变的染色质修饰基因之一,其中在肾透明细胞癌中有13%发生突变,并以截短突变(基因突变导致基因产物的缩短)为主。原发性ccRCC(肾透明性细胞癌)肿瘤的基因组图谱显示SETD2突变与转移呈正相关。然而,SETD2的丢失与否以及如何促进转移尚不清楚。因此,该研究主要利用转座酶和染色质可及性测序(ATAC-seq)、染色质免疫沉淀测序(ChIP-seq)和转录组测序(RNA-seq)进行综合多组学分析,建立了一个肿瘤抑制因子模型。在该模型中,SETD2介导的H3K36me3缺失,调节染色质的可及性,激活增强子,致癌基因转录被进一步的放大,最终促进肿瘤转移。此外,研究人员还发现了通过靶向特定的组蛋白伴侣复合物,比如ASF1 A/ASF1 B,为治疗SETD2缺陷型的肿瘤提供策略。
二、技术路线
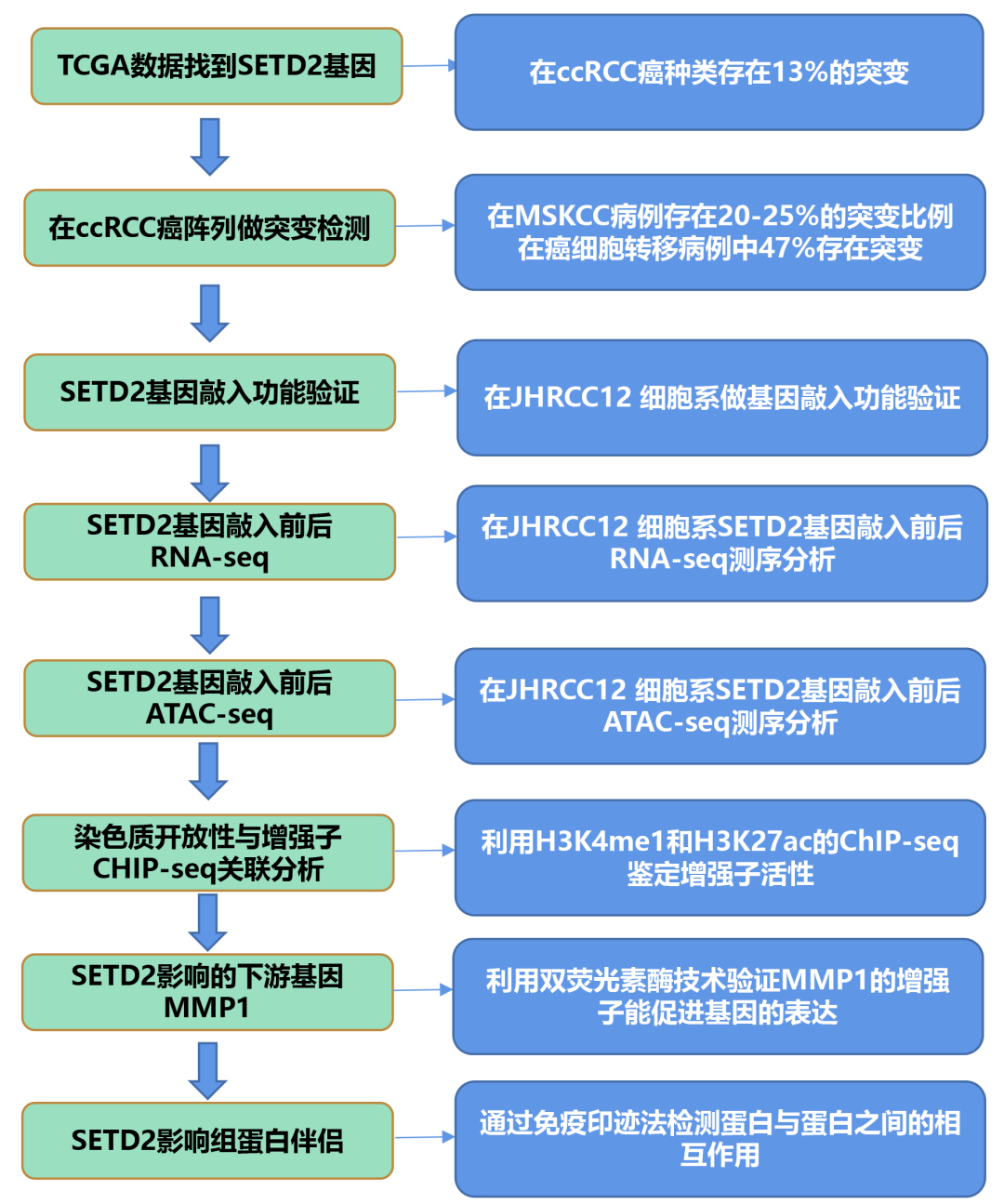
三、主要研究结论
1.在SETD2MT ccRCC中,H3K36me3的恢复可以抑制肿瘤转移。
2.SETD2缺失可上调ccRCC中的致癌转录程序。
3.SETD2缺失导致全基因组染色质可及性的增加。
4.SETD2的缺失诱导了表观遗传重构和增强子的激活。
5.SETD2缺失诱导MMP1表达促进ccRCC转移。
6.SETD2缺失增加了组蛋白伴侣向染色质的募集。
7.SETD2缺失使癌症对组蛋白伴侣的抑制敏感。
四、结果展示
1.SETD2缺失可上调ccRCC中的致癌转录程序,促进肿瘤转移。
为了评估SETD2突变与ccRCC转移的关系,首先研究人员收集了90例原发性和51例转移性ccRCC肿瘤患者,对595个癌症相关基因进行了靶向测序。此外,针对286例原发性ccRCC肿瘤进行了肿瘤基因检测(MSK-IMPACT)分析。发现原发性ccRCC肿瘤中的SETD2突变率从13%增加到20-25%,在转移性ccRCC肿瘤中SETD2的突变率增加到47%。
为了揭秘其潜在的分子机制,研究人员使用了携带突变的人转移性肿瘤来源的ccRCC细胞系(JHRCC12细胞系),构建免疫缺陷小鼠,建立原位异种移植模型。在亲代JHRCC12原位异种移植的15只小鼠中,超过90%的小鼠发生了胰腺转移,高达80%的小鼠发生了肝脏和膈肌转移。H3K36me3的恢复完全抑制了JHRCC12细胞的转移能力。SETD2∆N转导后H3K36me3的恢复大大减少了JHRCC12细胞向脑、肺和肝的转移。总之,这些数据表明SETD2介导的H3K36me3的缺失促进了肾癌的转移。
为了进一步研究JHRCC12细胞中H3K36me3缺失诱导的转录组变化。研究者使用RNA-seq技术进行分析,结果显示缺陷的H3K36me3中致癌通路发生富集,而H3K36me3恢复致癌通路下调。SETD2失活似乎进一步放大了由VHL和PBRM1联合缺失引起的致癌转录输出。SETD2介导的H3K36me3的缺失可以放大致癌转录输出,从而促进ccRCC的转移。
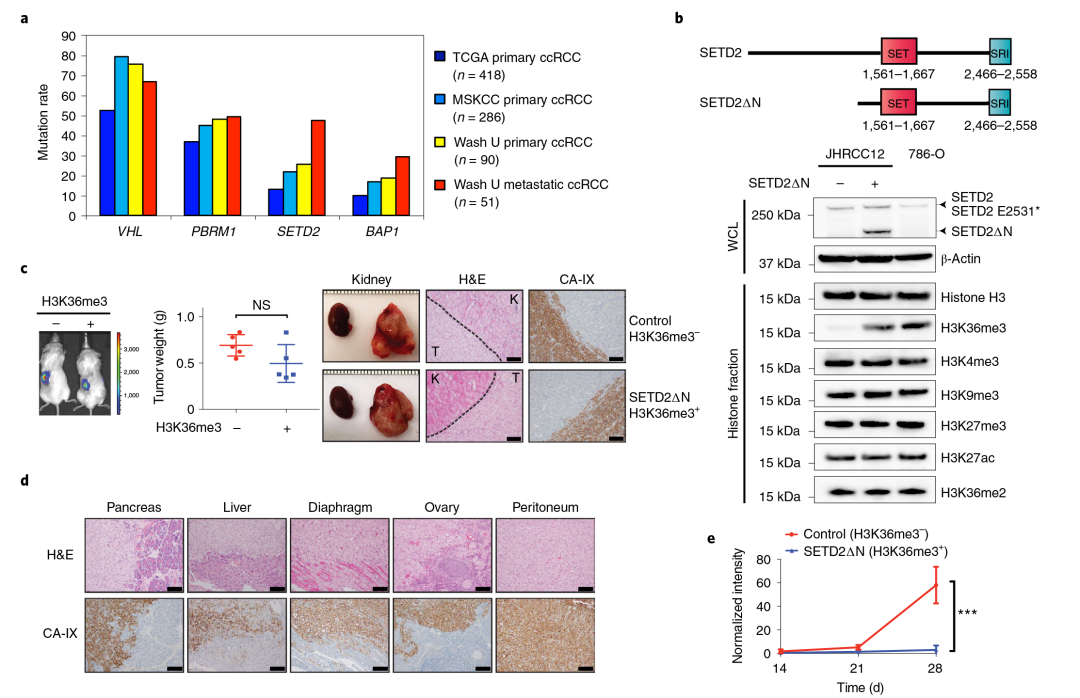
图1 SETD2在转移性ccRCC肿瘤中高度突变,并且SETD2MT ccRCC中H3K36me3的恢复抑制肿瘤转移。(a)VHL、PBRM1、SETD2和BAP1在上述ccRCC队列中的突变率;(b)SETD2和SETD2ΔN的结构域结构示意图;(c)将荧光素酶转导的JHRCC12细胞注射到NOD的单侧肾肾下囊中;(d)JHRCC12细胞的小鼠转移性肿瘤器官CA-IX的代表性HE染色和IHC染色;(e)在心内注射荧光素酶转导的JHRCC12细胞后,无胸腺裸鼠的生物发光图像。
2.SETD2缺失导致全基因组染色质可及性的增加。
为了确定SETD2MT ccRCC中致癌通路转录输出的增加是否是由H3K36me3缺失后表观遗传景观的改变引起的,在JHRCC12细胞中进行ATAC-seq检测,以评估H3K36me3对染色质可及性的影响。结果显示,在显著改变的peaks中,90.8%的峰在H3K36me3缺失后染色质可及性增加,ATAC-seq peaks(FDR<0.05;占总peaks的30.5%)中36.1%位于内含子,50.3%在基因间区,12.4%位于启动子,1.2%位于外显子。同时在SETD2缺陷的RTE细胞(原代肾小管上皮细胞)和ccRCC人来源的异种移植模型中也观察到染色质可及性的全基因组增加。
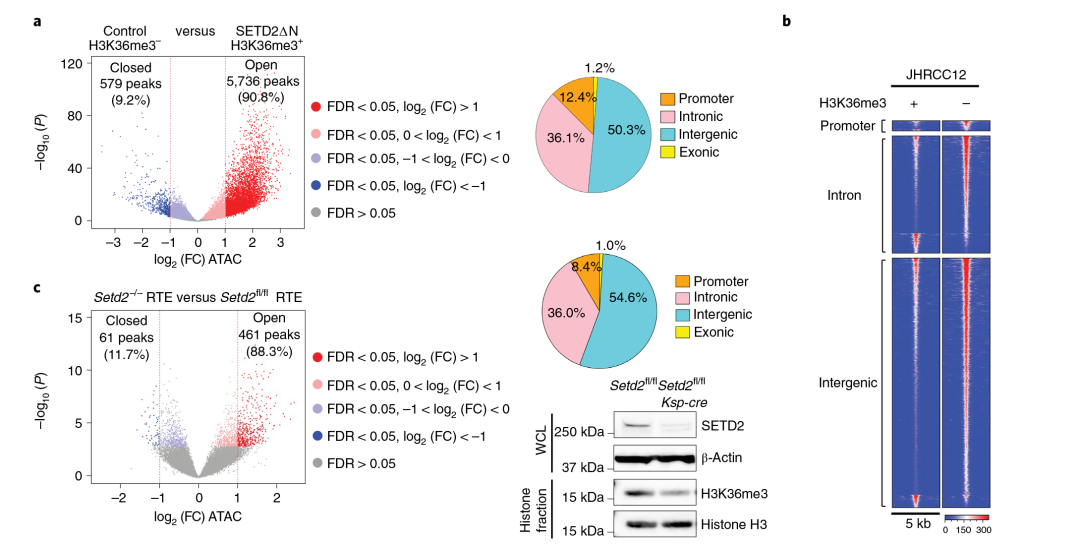
图2 SETD2介导的H3K36me3的缺失诱导染色质可及性的全基因组增加。(a)对照(H3K36me3-)与SETD2∆N转导(H3K36me3+)JHRCC12细胞的ATAC-seq peaks火山图;(b)ATAC-seq peaks在启动子、内含子和基因间区域的定位分组;(c)比较SETD2fl/flKsp-cre+小鼠与SETD2fl/fl小鼠培养的原代小鼠RTE细胞的ATAC-seq峰的火山图。
3.SETD2介导的H3K36me3缺失增加了染色质可及性,进而增强肾癌的致癌转录输出。
通过整合ATAC-seq数据和RNA-seq数据,发现在H3K36me3缺失后,在开放染色质区域发现了STAT家族转录因子结合基序的富集,这与富集的STAT转录特征相一致。总的来说,数据显示SETD2介导的H3K36me3缺失增加了染色质可及性,进而增强肾癌的致癌转录输出。
4.SETD2的缺失诱导了表观遗传重构和增强子的激活。
鉴于H3K36me3缺失诱导的开放染色质主要集中在包含增强子的内含子和基因间区域,研究人员对H3K36me3缺陷的JHRCC12细胞进行增强子标记H3K4me1和H3K27ac,以及启动子标记H3K4me3和抑制性标记的H3K27me3进行ChIP-seq检测。需要注意的是,大多数差异富集的增强子在H3K36me3缺失时增加,在H3K36me3缺失的亲本JHRCC12细胞中,高达93.2%的H3K4me1差异峰和69.3%的H3K27ac差异峰显示信号增加。此外,ATAC-seq与H3K4me1的ChIP-seq(皮尔逊相关系数=0.88)和H3K27ac的ChIP-seq(皮尔逊相关系数=0.80)的变化呈显著的正相关。也就是说H3K36me3缺失增加染色质可及性,进而激活增强子,SETD2介导的H3K36me3缺失通过增加染色质的可及性,激活增强子进而放大致癌转录输出。
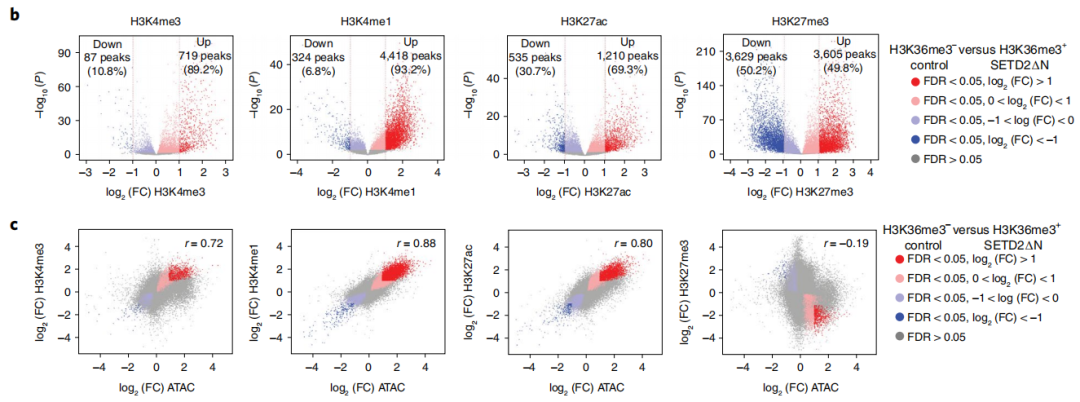
图3 SETD2介导的H3K36me3的缺失诱导全基因组表观遗传重编程和增强子的激活。(b)火山图显示了H3K36me3−与H3K36me3+ JHRCC12细胞的组蛋白修饰ChIP-seq变化;(c)散点图显示了H3K36me3−和H3K36seq每个组蛋白修饰的log2(FC)和H3K36me3+ JHRCC12细胞的ATAC-seq的log2(FC)之间的相关性。
5.SETD2缺失诱导MMP1表达促进ccRCC转移。
为了验证SETD2缺失介导的H3K36me3增加染色质可及性激活增强子,如何诱导基因参与转移?研究人员选择了一个肿瘤转移促进基因MMP1,通过CHIP-qPCR技术进行检测。结果表明,SETD2介导的H3K36me3的缺失增加了MMP1的染色质可及性和内含子增强子活性,从而诱导其表达,最终有助于肾癌的转移。
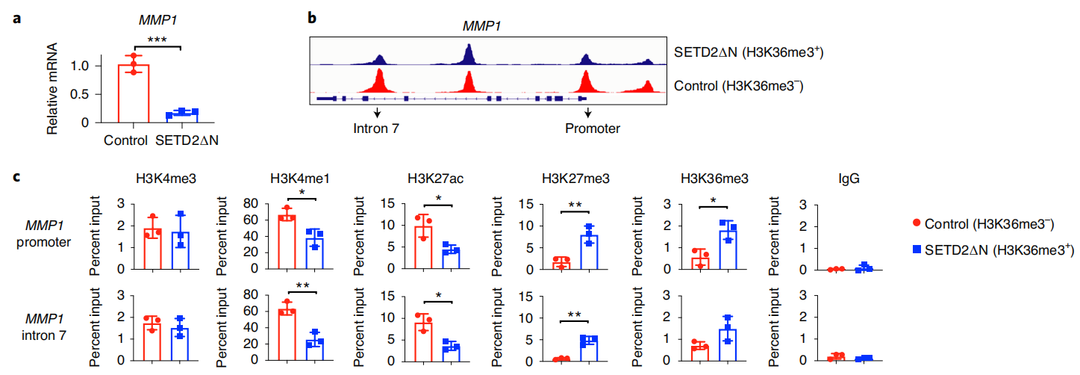
图4 SETD2功能缺失诱导MMP1表达促进ccRCC转移。(a) RT-qPCR检测JHRCC12细胞中的MMP1 mRNA;(b)ATAC-seq在JHRCC12细胞的MMP1位点;(c)用ChIP-qPCR检测JHRCC12细胞中MMP1的启动子和第7内含子。
6.SETD2缺失增加了组蛋白伴侣向染色质的募集,使癌症对组蛋白伴侣的抑制敏感
组蛋白伴侣调节核小体的组装和拆卸,并控制染色质动力学。在酵母中,Set2(SETD2的同源物)介导的H3K36me3已被证明可以抑制H3与组蛋白伴侣Asf1和Spt16的相互作用,阻止组蛋白在编码区的交换,并降低基因体中Pol II的可及性。因此,研究人员推测H3K36me3缺失诱导的染色质可及性的增加可能是由组蛋白伴侣的异常招募引起的。该研究发现,在JHRCC12细胞中H3K36me3的恢复大大降低了核和染色质部分的ASF1 A和ASF1 B(Asf1的人类同源物),而没有降低SPT16(Spt16的人类同源物)。Set2缺失诱导的Asf1染色质募集的增加会导致H3K56ac的富集,这是由于Asf1介导的H3K56ac交换。在JHRCC12细胞中H3K36me3的缺失是否会影响组蛋白交换,从而增加H3K56ac的沉积。ChIP-seq显示,大多数差异富集的H3K56ac峰(FDR<0.05)在H3K36me3丢失后增加。H3K56ac的变化与JHRCC12细胞中H3K36me3缺失诱导的染色质可及性的改变呈正相关。总的来说,SETD2介导的H3K36me3缺失与ASF1 A/ASF1 B和H3K56ac沉积的核/染色质关联增加相关。
如果H3K36me3缺失诱导的组蛋白交换增加需要创建一个允许的表观遗传景观来增强致癌转录输出,那么可以想象SETD2缺陷的癌症对ASF1 A/ASF1 B失活更敏感。研究者发现与H3K36me3缺陷的JHRCC12细胞相比,H3K36me3正常表达的JHRCC12细胞诱导了更多的凋亡。相反,使用两种独立的sgRNAs,SETD2的KO使CAKI-2细胞在ASF1 A和ASF1 B的KO后发生凋亡。虽然H3K36me3的缺失对SPT16染色质关联的影响很小,但SETD2功能的缺失使JHRCC12和CAKI-2细胞对由SUPT16H(SPT16基因)的KO引发的凋亡敏感。H3K36me3缺失诱导的ASF1A/ASF1B染色质募集可能导致染色质对包括SPT16在内的其他组蛋白伴侣的可及性增加,建立一个前馈扩增环来维持开放染色质状态。
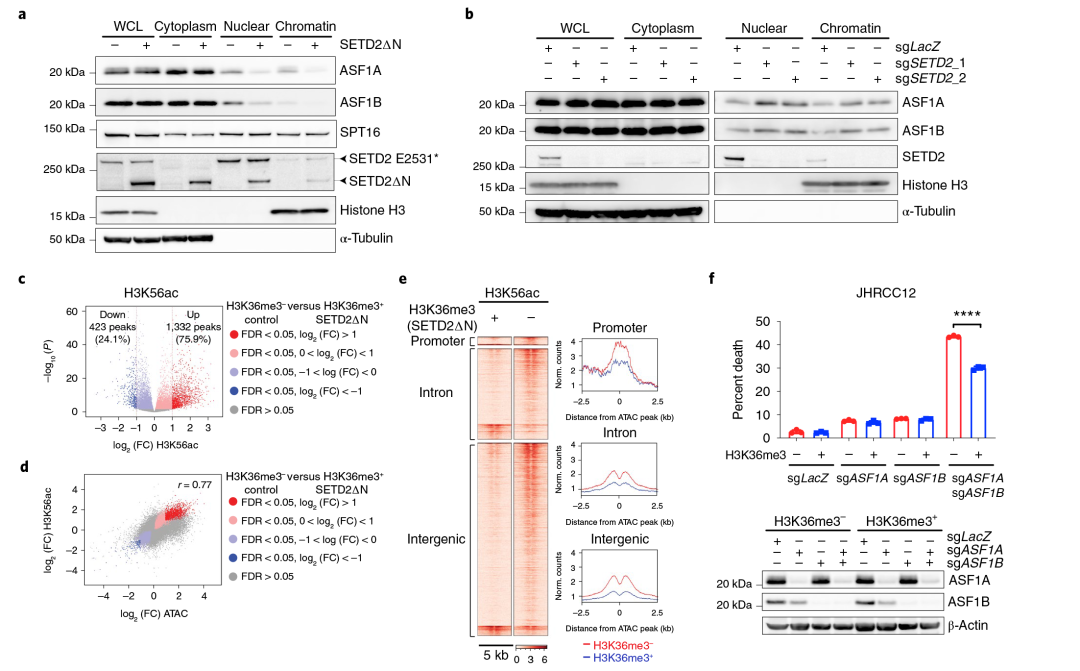
图5 SETD2介导的H3K36me3缺失增加组蛋白伴侣募集并使癌症细胞对组蛋白基因的失活敏感。(a)用免疫印迹法分析了JHRCC12细胞的WCLs和细胞质、细胞核和染色质组分;(b)用免疫印迹法分析了CAKI-2细胞的WCL和细胞质、细胞核和染色质组分;(c)火山图显示对照(H3K36me3−)和SETD2∆N转导(H3K36me3+)JHRCC12细胞中H3K56ac的ChIP-seq的变化;(d)H3K36me3−与H3K36me3+ JHRCC12细胞中H3K56ac ChIP-seq的log2(FC)和ATAC-seq的log2(FC)之间的相关性;(e)热图和超峰图显示H3K56ac ChIP-seq在ATAC-seq峰的相同区域的5kb窗口;(f)H3K36me3−和H3K36me3+(SETD2ΔN转导的)JHRCC12细胞经慢病毒CRISPR/cas9介导的ASF1 A(第1外显子)、ASF1 B或ASF1 A(第1外显子)和ASF1 B,并通过免疫印迹分析。
五、小结
该研究通过对RNA-seq、ATAC-seq和ChIP-seq数据的综合分析揭示了SETD2的表观遗传肿瘤抑制模型,其中SETD2介导的H3K36me3缺失创建了一个由开放染色质结构和活性/许可组蛋白修饰组成的表观遗传景观,使协同致癌驱动因素能够进一步放大转录输出。可以想象,SETD2缺失后激活的致癌转录程序不仅依赖于各自的功能驱动因素,还依赖于不同癌症类型中参与的转录因子和染色质修饰因子。这有助于解释为什么SETD2突变发生在多种人类癌症中,并与多种致癌驱动因素相关。
研究结果表明,SETD2介导的H3K36me3缺失增加了转录输出。SETD2介导的H3K36共转录甲基化似乎通过减少组蛋白伴侣对组蛋白和限制组蛋白的结合亲和力来维持封闭的染色质结构。表观遗传调控因子作为一种新的主要癌症基因的发现,为新的癌症疗法的开发带来了挑战和机遇。在阐明相关肿瘤转移机制的基础上,为SETD2缺陷癌症的治疗提供治疗靶点。鉴于SETD2在多种人类癌症中都发生了突变,因此该研究将为癌症生物学和肾癌以外的治疗方法提供见解。
原文链接:
https://www.nature.com/articles/s43018-021-00316-3

















 832
832

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









