



 Seqkit是一款用Go语言编写的,适用于fasta/q序列文件处理的软件。它支持多平台,无需依赖,运行快速,提供包括Seq、subseq、translate、concat等多种命令,方便进行序列操作和数据分析。
Seqkit是一款用Go语言编写的,适用于fasta/q序列文件处理的软件。它支持多平台,无需依赖,运行快速,提供包括Seq、subseq、translate、concat等多种命令,方便进行序列操作和数据分析。
大家是否还在为查看连接多个序列文件而感到烦恼呢?是否还在为查找某一段碱基而感到焦虑呢?别担心,有了这款软件这些问题就会自然而然的解决啦,这款软件是什么呢,请让小编为你一一介绍吧~
软件介绍
Seqkit是一款专门处理fsata/q序列文件的软件,由go语言编写,功能比较完善,软件使用也很稳定。
优点
1.能够非常全面的处理fasta/q文件,运行速度超快的序列工具
2.支持多平台(Linux/Windows/macOS)使用,是一款轻量级软件
3.可以做到开箱即用(无依赖,无需编译,无需配置)
安装方法
方法一:下载二进制文件(最新的稳定/开发版本)
下载地址:https://bioinf.shenwei.me/seqkit/download/只需要载您的操作系统的压缩可执行文件,并使用tar -zxvf *.tar.gz命令或其他工具解压即可
方法二:通过conda安装(最新稳定版)
conda install -c bioconda seqkit
方法三:通过homebrew安装(最新稳定版)
brew install seqkit
方法四:对于 Go 开发者(最新的稳定/开发版本)
go get -u github.com/shenwei356/seqkit/seqkit
方法五:基于 Docker 的安装(最新的 stable/dev 版本)
安装 Docker
# git clone 这个仓库:
git clone https://github.com/shenwei356/seqkit
运行以下命令:
cd seqkit
docker build -t shenwei356/seqkit .
docker run -it shenwei356/seqkit:latest
参数
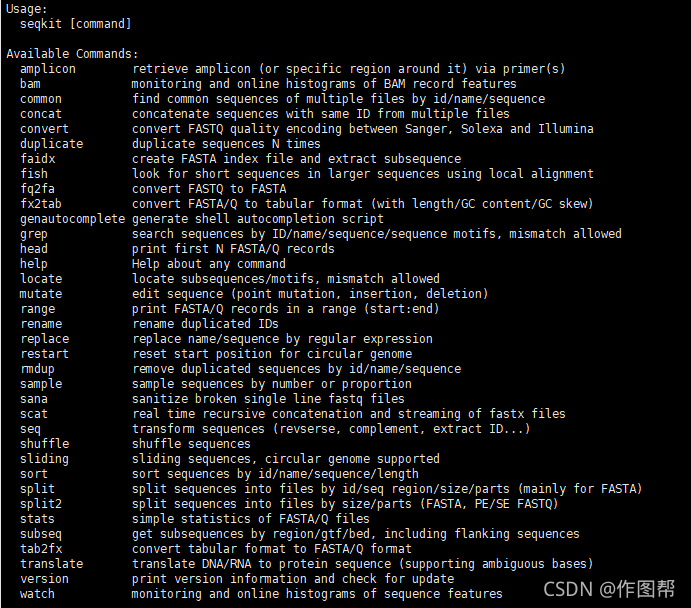
常用命令
1.Seq
$ seqkit seq hairpin.fa.gz #查看 fa文件
$ seqkit seq reads_1.fq.gz #查看fq文件
$ seqkit seq hairpin.fa.gz -n #打印序列全名
$ seqkit seq hairpin.fa.gz -n -i #打印序列名的唯一标识
$ seqkit seq hairpin.fa.gz -n -i --id-regexp "^[^\s]+\s([^\s]+)\s"#打印ID中的第二个字段
$ seqkit seq hairpin.fa.gz -s -w 0 #只打印序列(-w定义输出行宽,0不换行,默认为60)
$ seqkit seq hairpin.fa.gz -r -p #反向互补(-r 序列反向;-p序列互补)
$ echo -e ">seq\nACGT-ACTGC-ACC" | seqkit seq -g -u #删除gap并大写碱基(-g 去除序列中的间隔;-u转化序列为大写字母展示)
>seq
ACGTACTGCACC
$ echo -e ">seq\nUCAUAUGCUUGUCUCAAAGAUUA" | seqkit seq --rna2dna #RNA 转DNA
>seq
TCATATGCTTGTCTCAAAGATTA
2.subseq 获取
建议:使用普通的FASTA文件,这样seqkit就可以使用FASTA索引
$ zcat hairpin.fa.gz | seqkit subseq -r 1:12 #提取前 12 个碱基
$ zcat hairpin.fa.gz | seqkit subseq -r -12:-1 #提取后 12 个碱基
$ zcat hairpin.fa.gz | seqkit subseq -r 13:-13#提取第13到倒数第13个,即去掉前12个和后12个
3.translate 翻译DNA/RNA为蛋白质序列
$ seqkit translate genome.fa |head #转化为蛋白序列
$ seqkit translate genome.fa --trim | head #去除”*”
4.concat 连接
$ seqkit concat <(seqkit subseq -r 1:2 t.fa) <(seqkit subseq -r -2:-1 t.fa)#连接前面两个碱基和后面两个碱基
$ seqkit concat 1.fa 2.fa #将两个fa文件合二为
5.stats 数据统计
$ seqkit stats *.f{a,q}.gz #统计序列信息
$ seqkit stats *.f{a,q}.gz -T #用tab分割
$ seqkit stats *.f{a,q}.gz -a # 统计更多信息
$ seqkit stats -j 10 refseq/virual/*.fna.gz #多文件统计( -j:使用多线程)
6.faidx 建立索引文件、提取子序列
$ seqkit faidx hairpin.fa #建立序列索引
$ seqkit faidx tests/hairpin.fa hsa-let-7a-1 hsa-let-7a-2 #提取ID信息
$ seqkit faidx tests/hairpin.fa hsa-let-7a-1 hsa-let-7a-2 -f # -f 标题全部输出
$ seqkit faidx tests/hairpin.fa hsa-let-7a-1:1-10 #提取子序列第1个到第10个碱基
$ seqkit faidx tests/hairpin.fa hsa-let-7a-1:-10--1 #提取子序列最后10个碱基
$ seqkit faidx tests/hairpin.fa hsa-let-7a-1:1 #提取子序列第1个碱基
7.fa2fa fa文件转换为fa文件
$ seqkit fq2fa reads_1.fq.gz -o reads_1.fa.gz #fq转fa
8.fx2tab & tab2fx 将fasta/q转换为表格形式
$ seqkit fx2tab hairpin.fa.gz | head -n 2 #序列转化表格格式
$ seqkit fx2tab hairpin.fa.gz -l -g -n -i -H | head #打印序列长度、GC含量
(注释:-l 统计序列长度;-g 统计平均GC含量;-i 只打印名称(不打印序列);-H 打印标题行)
$ zcat hairpin.fa.gz | seqkit fx2tab | seqkit tab2fx #表格转序列形式
#转为表格后排序,再转换回序列(以下两种等同)
$ zcat hairpin.fa.gz \
| seqkit fx2tab -l \
| sort -t"`echo -e '\t'`" -n -k4,4 \
| seqkit tab2fx
$ seqkit sort -l hairpin.fa.gz
9.sort 排序
$ echo -e ">seq1\nACGTNcccc\n>SEQ2\nacgtnAAAA" | seqkit sort --quiet#按ID排序
$ echo -e ">seq1\nACGTNcccc\n>SEQ2\nacgtnAAAA" | seqkit sort --quiet -i #按 ID 排序,忽略大小写
$ echo -e ">seq1\nACGTNcccc\n>SEQ2\nacgtnAAAA" | seqkit sort --quiet -s -i #按 seq 排序,忽略大小写
$ echo -e ">seq1\nACGTNcccc\n>SEQ2\nacgtnAAAAnnn\n>seq3\nacgt" | seqkit sort --quiet -l#按序列长度排序
10.grep 匹配
$ seqkit grep -f id.txt seqs.fq.gz -o result.fq.gz#按 ID 列表文件搜索(不包含空格)
$ seqkit grep -i -f id.txt seqs.fq.gz -o result.fq.gz #-i 忽略大小写
$ seqkit grep -n -f name.txt seqs.fa.gz -o result.fa.gz #使用序列名称列表进行搜索(它们可能包含空格)
$ cat hairpin.fa.gz | seqkit grep -s -i -p aggcg #提取包含 AGGCG 的序列
$ zcat hairpin.fa.gz | seqkit grep -s -r -i -p ^aggcg #提取以 AGGCG 开头的序列
11.spilt 拆分
$ seqkit split hairpin.fa.gz -s 10000 #将序列拆分为最多10000个序列的部分
$ seqkit split hairpin.fa.gz -p 4 #将序列拆分为4部分
$ seqkit split hairpin.fa.gz -p 4 -2#加上-2减少内存使用
$ seqkit split hairpin.fa.gz -i --id-regexp "^([\w]+)\-" -2 #按id拆分序列
$ seqkit split hairpin.fa.gz -r 1:3 -2 #按前三个序列碱基来区分
上述常用命令,有一些使用方法如下图:
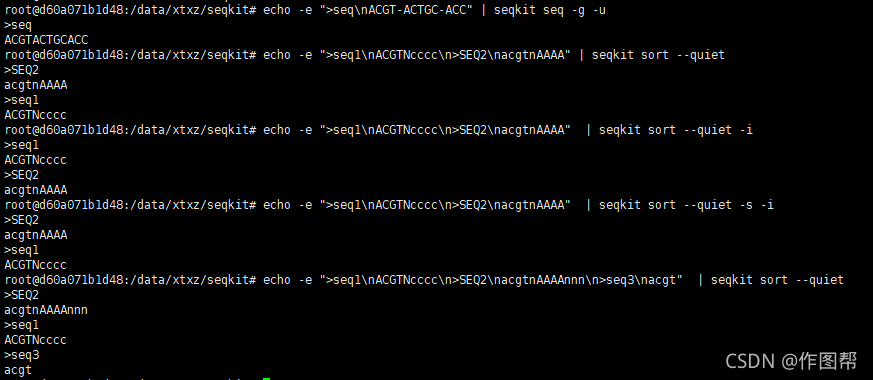
仅供参考哦!还有一些命令还需大家自行挖掘啦,用法介绍的参考地址是https://bioinf.shenwei.me/seqkit/usage,以上就是关于序列处理工具-Seqkit的介绍啦! 有问题可以联系图图哦~


















 6846
6846

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









