




 CD-HIT是一款用于去除冗余序列的高效软件,尤其适用于蛋白质聚类。其工作原理基于序列相似性排序和word过滤,显著提高了处理速度。安装后,通过指定参数如相似性阈值(默认0.9)即可完成去冗余操作。此外,文中还提及了pandas在数据分析中的基础应用。
CD-HIT是一款用于去除冗余序列的高效软件,尤其适用于蛋白质聚类。其工作原理基于序列相似性排序和word过滤,显著提高了处理速度。安装后,通过指定参数如相似性阈值(默认0.9)即可完成去冗余操作。此外,文中还提及了pandas在数据分析中的基础应用。
cd-hit 去除冗余序列
CD-HIT早先是一个蛋白聚类的软件,其主要的特定就是快!
其去除冗余序列的大概思路就是:
首先对所有序列长度进行排序,从最长的开始,形成一个序列类,然后依次对序列进行处理,如果下一条序列和代表的序列相似性在cutoff,则将其加入同一类,否则得到新的类。
所以快主要是两个方面的原因:一个是使用了word过滤方法,即如果两条序列之间的相似性在80%(假设序列长度为100),那么它们至少有60个相同的长度为2的word,至少有40个相同的长度为3的word,至少有20个相同的长度为4的word。基于这个原则,在处理新的序列的时候,如果新的序列与已有序列的相同word的长度不能满足这些要求则不需要进行比对了,这极大的降低了时间消耗;另外一个速度快的原因是使用了index table,可以很快的计算序列之间相同word的数目。
当序列相似性在80%时,有20个位点是有差异的,极端的情况就是这20个位点对应的长度为2的字符串都不一样,因此是40个不一样,当有更多的不一样时,两条序列的相似性不可能在80%;同理,如果这20个位点对应的长度为4的字符串都不一样,则有80个不一样。
安装
点击该处进行下载https://github.com/weizhongli/cdhit/archive/V4.6.2.tar.gz
tar -zxf cdhit-4.6.2.tar.gz
cd tar -zxf cdhit-4.6.2.tar.gz
make
其输入格式为fasta的格式
简单使用
cd-hit -i all.fasta -o new.fa -c 0.9 -aS 0.8 -d 0
参数
-i : 输入文件,fasta
-o: 输出文件
-c: 类似比对相似性高于0.9 则聚为一类,一般为0.9
-d: 当为0时,利用fasta文件完整名称
还有其它一些参数,可以利用下面进行理解
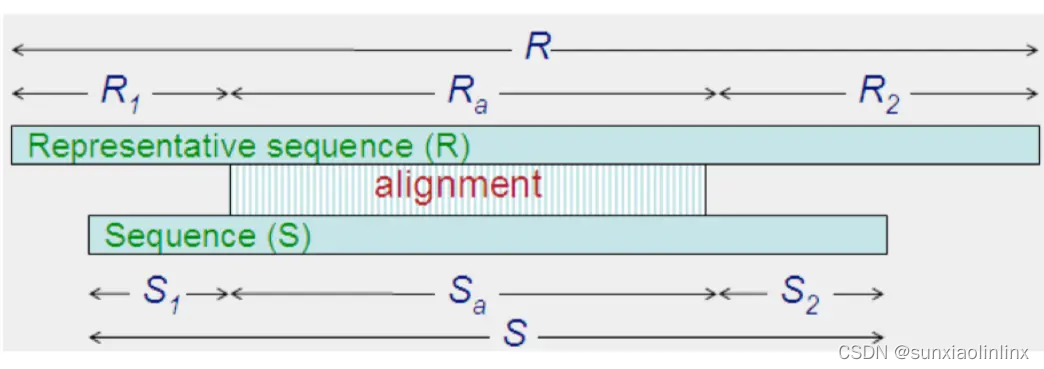
aL=Ra /R
AL = R - Ra
aS=Sa /S
AS = S - Sa
s=Sa /Ra
S=R/S
U=S1 +S2
uL = U / R
uS = U / S
输出
两个结果
去冗余后的fasta文件
说明文档,其内容大概如下

其中“>” cluster, ‘*’ 该序列是这个cluster的代表序列,‘%’,相似性
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1508
1508

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









