



1. conda 环境建立
conda create -n atac_test
conda activate atac_test
2.raw data 进行质控 QC (fastqc / fastp )
# 单一运行
fastqc ck_1.fastq.gz
fastqc ck_2.fastq.gz
# 指定输出目录和读取目录
fastqc -o ./raw_fastqc -t 4 ./FASTQ/*.fastq.gz #-o 输出目录 -t 4 进程数 读取目录
输出QC质控网页报告
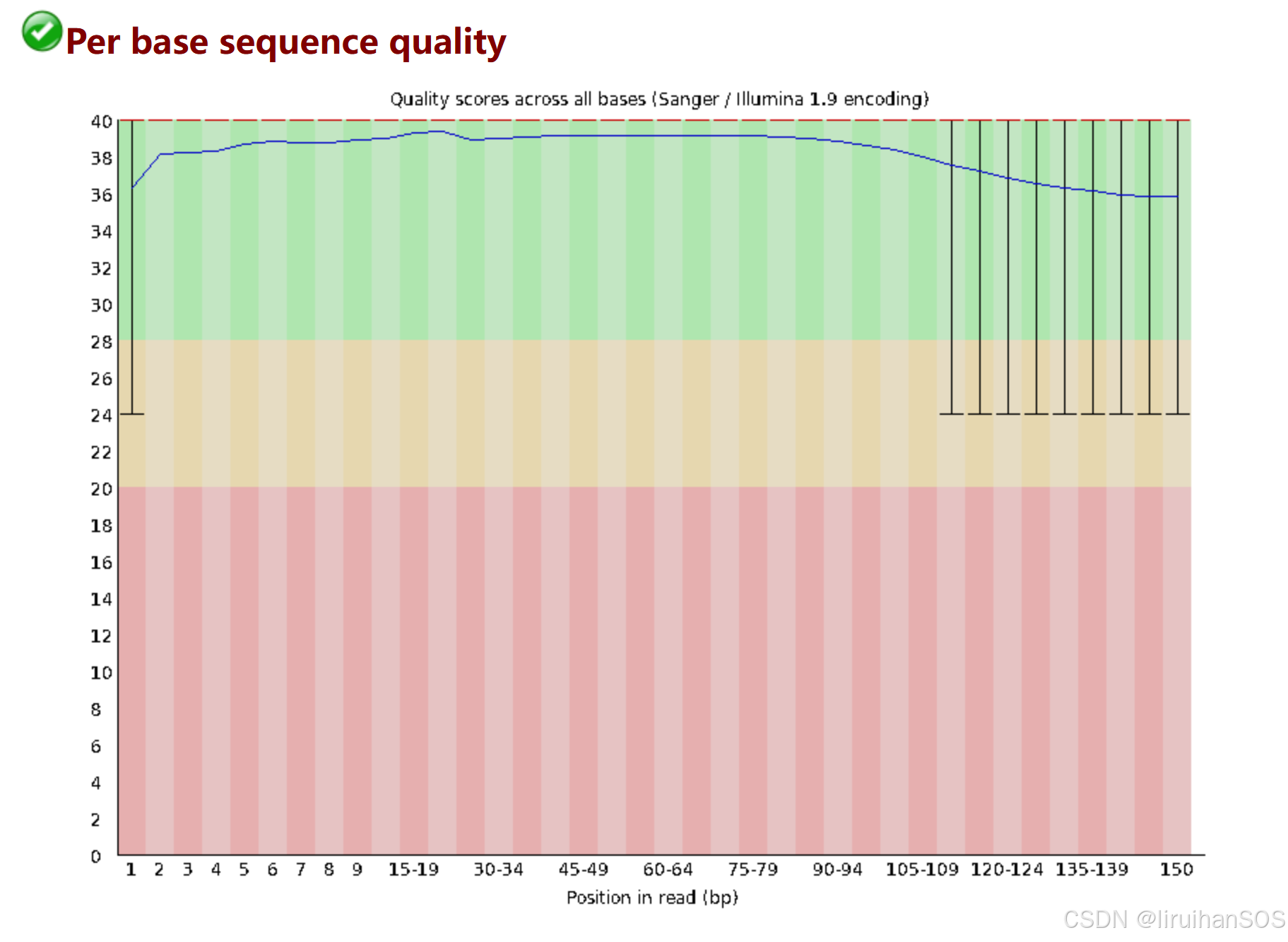
3. raw data 过滤数据得到 clean data (fastp 和 trim_galore 二选一 )
# Fastp PE双端
fastp\
--i ck_1.fq.gz \ # read1原始fq文件
-I ck_2.fq.gz \ # read2原始fq文件
-o ck_clean_1.fq.gz \ # read1过滤后输出的fq文件
-O ck_clean_2.fq.gz \ # read2过滤后输出的fq文件
--detect_adapter_for_pe \
-q 20 -u 40 \
--thread 10 \
--trim_poly_x \
-h ck_report.html -j ck_report.json
# trim_galore PE双端
trim_galore --paired --phred33 --length 35 -q 30 \ # q30
--fastqc ck_1.fq.gz ck_2.fq.gz -o ./cleaned #输出文件夹
fastp 默认启用质量过滤、接头识别、长度过滤等基础功能
trim_galore :GitHub - FelixKrueger/TrimGalore:Cutadapt 和 FastQC 的包装器,用于一致地对 FastQ 文件应用适配器和质量修剪,并为 RRBS 数据提供额外的功能
trim_galore 包含 Cutadapt 和 FastQC ,运行会返回总结结果
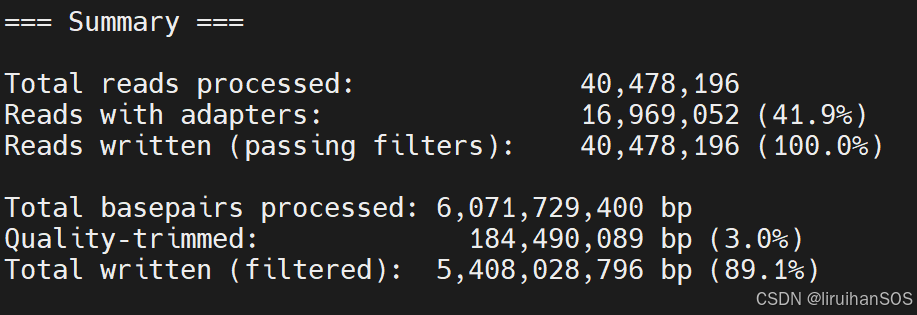
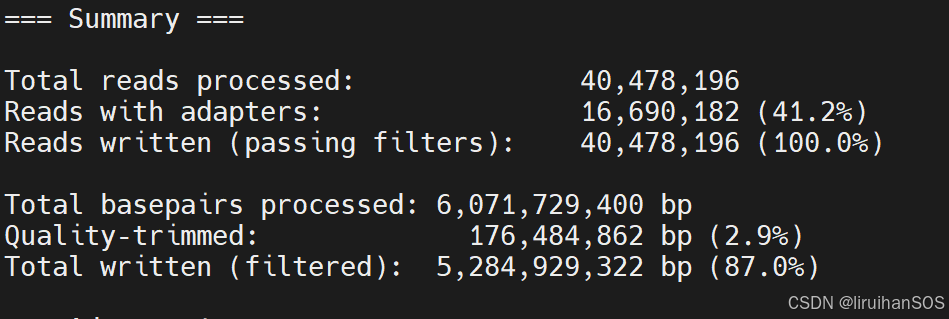
生成文件:

Validated Data:在修剪基础上,经过严格验证和过滤的高质量双端数据,确保下游分析的可靠性。
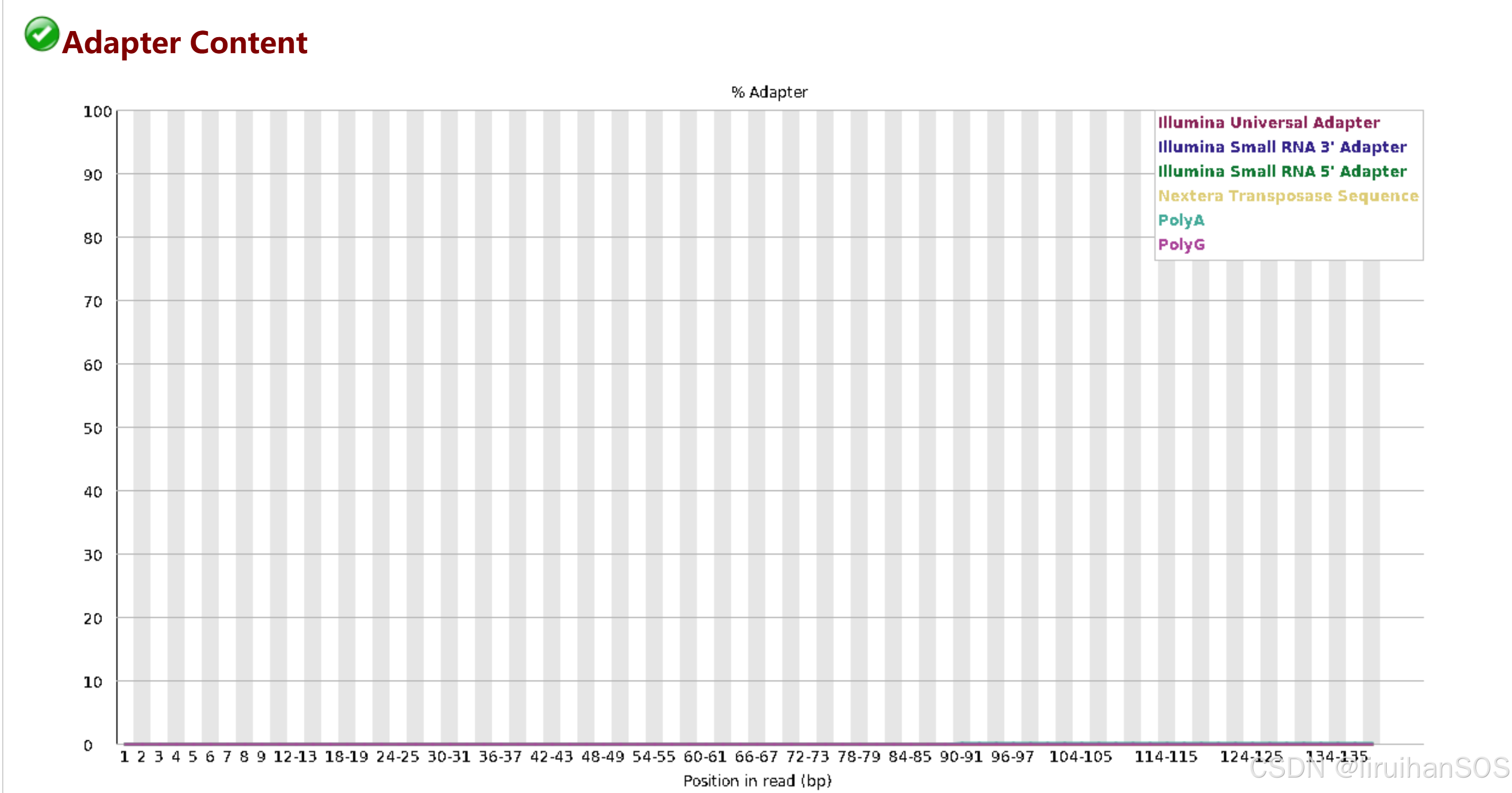
trim 和 validation 后会fastqc validation的序列文件 。
再次检查修剪和验证后的QC结果:
4. 序列比对
下载hg38 基因组文件:
wget http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz
gunzip hg38.fa.gz
构建基因组的bowtie2索引文件
当时因为安装libstdc++.so.6缺少CXXABI_1.3.11版本,所以单独创建了一个指定libstdc 版本的conda环境
conda create -n env_bowtie2 libstdcxx-ng=9.3.0 tbb=2020.2
conda activate env_bowtie2
conda install bowtie2bowtie2-build reference_genome.fa genome_index reference_genome.fa 替换成样本对应的参考基因组文件,生成6个文件。(时间有点长)
Bowtie2 比对: (注意输入文件的正反斜杠 )
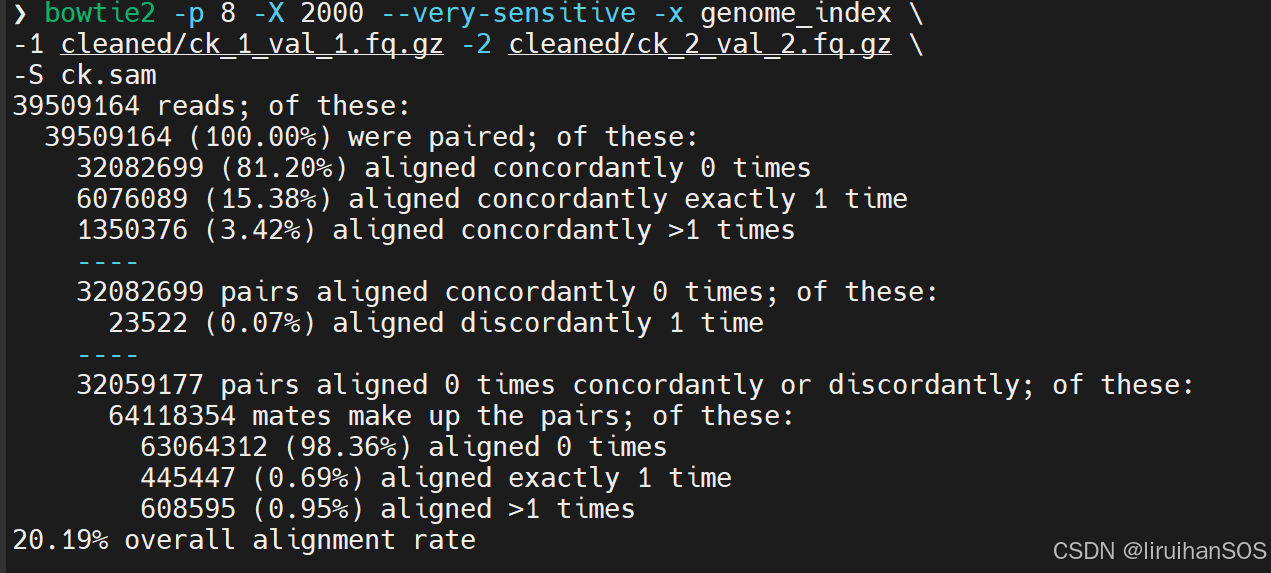
bowtie2 -p 8 -X 2000 --very-sensitive -x genome_index \
-1 cleaned/ck_1_val_1.fq.gz -2 cleaned/ck_2_val_2.fq.gz \
-S ck.sam-X 2000 参数: 设置双端测序数据的最大插入片段长度(即左右两端reads之间的最大间隔)是2000bp(参考自 2013年 ATAC nature methods :Reads were aligned to hg19 using Bowtie with the parameters -X2000 and -m1)
5.sam 转换成 bam 并排序
samtools view -bS ck.sam | samtools sort -@ 4 -o ck_sorted.bam
samtools index ck_sorted.bamsam 是文本格式,bam 是二进制格式,bam 比 sam 格式存储和读写更快,方便后续处理、建立索引和IGV可视化。
6. 过滤低质量的比对
①保留比对质量较高的q30 (MAPQ ≥ 30)
-b:输出为 BAM 格式(默认是 SAM 格式)。
-F 4:过滤掉未比对的 reads(FLAG 值为 4 的 reads)。
ck_sorted.bam:输入的未过滤的 BAM 文件。
-o filtered_ck.bam:指定输出文件名。
samtools view -b -F 4 -q 30 ck_sorted.bam -o filtered_ck.bam |
samtools view -c filtered_ck.bam # 统计比对的reads 数
②去除PCR重复
工具选择: picard (java)、 sambamba 处理大量文件时速度更快
# 因为java 版本不够17 ,用不了 3.3.0 版本的 Picard ,需强制安装java17
conda install -c conda-forge openjdk=17
# 下载Picard
wget https://github.com/broadinstitute/picard/releases/download/3.3.0/picard.jar
# 验证安装是否成功
java -jar picard.jar输出下面内容就是安装成功了
#需要先创立group信息后再去除重复
java -jar picard.jar AddOrReplaceReadGroups \
-I 1_sorted.bam \
-O 1_sorted_withRG.bam \
--RGID Sample1 --RGLB Library1 --RGPL ILLUMINA --RGPU Unit1 --RGSM Sample1 --CREATE_INDEX true
java -jar picard.jar MarkDuplicates \
-I 1_sorted_withRG.bam \
-O 1_dedup.bam \
--METRICS_FILE duplicate_metrics.txt --REMOVE_DUPLICATES true --CREATE_INDEX true
sambamba 去除PCR重复:
conda install bioconda::sambamba
sambamba markdup -r -t 4 filtered_ck.bam ck_pcr.bam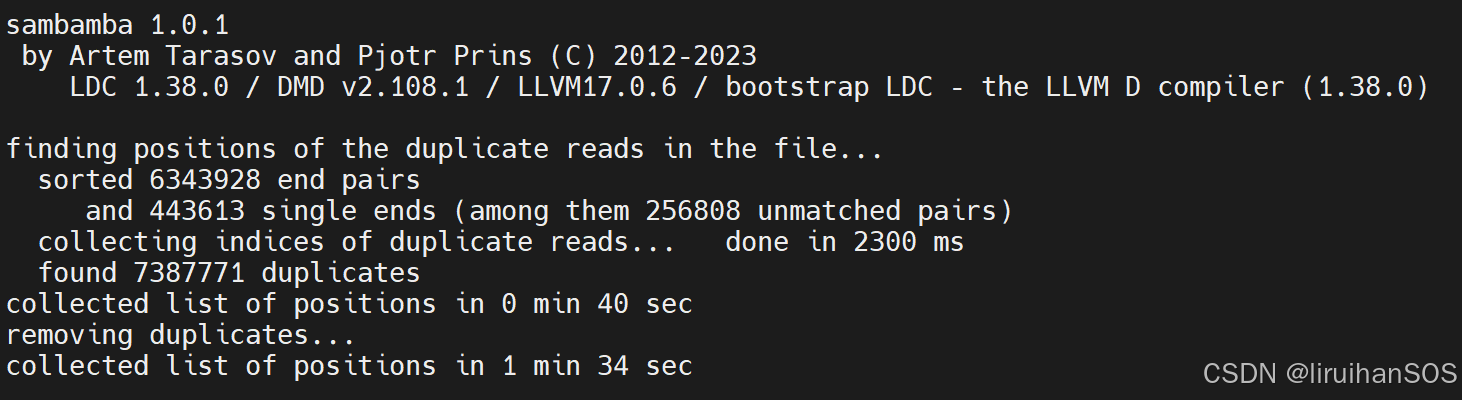
③过滤线粒体reads
# 1. 创建索引(生成ck_pcr.bam.bai)
samtools index ck_pcr.bam
# 2. 统计染色体比对信息(输出到ck_pcr.mitochondrial.stats)
samtools idxstats ck_pcr.bam > ck_pcr.mitochondrial.stats
# 3. 过滤线粒体chrM的reads,生成最终文件
samtools view -h ck_pcr.bam | grep -v 'chrM' | samtools view -bS -o ck.final.bam5. peak calling 和注释
ATAC-seq关心的是在哪切断,断点才是peak的中心,所以使用shift模型,–shift -75或-100
对人细胞系ATAC-seq 数据call peak的参数设置如下:(参考:ATAC-Seq分析教程:用MACS2软件call peaks | Public Library of Bioinformatics)
macs2 callpeak -t ck.final.bam -n sample --shift -100 --extsize 200 --nomodel -B --SPMR -g hs --outdir Macs2_out 2> ck.macs2.log


















 1万+
1万+


 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言








