




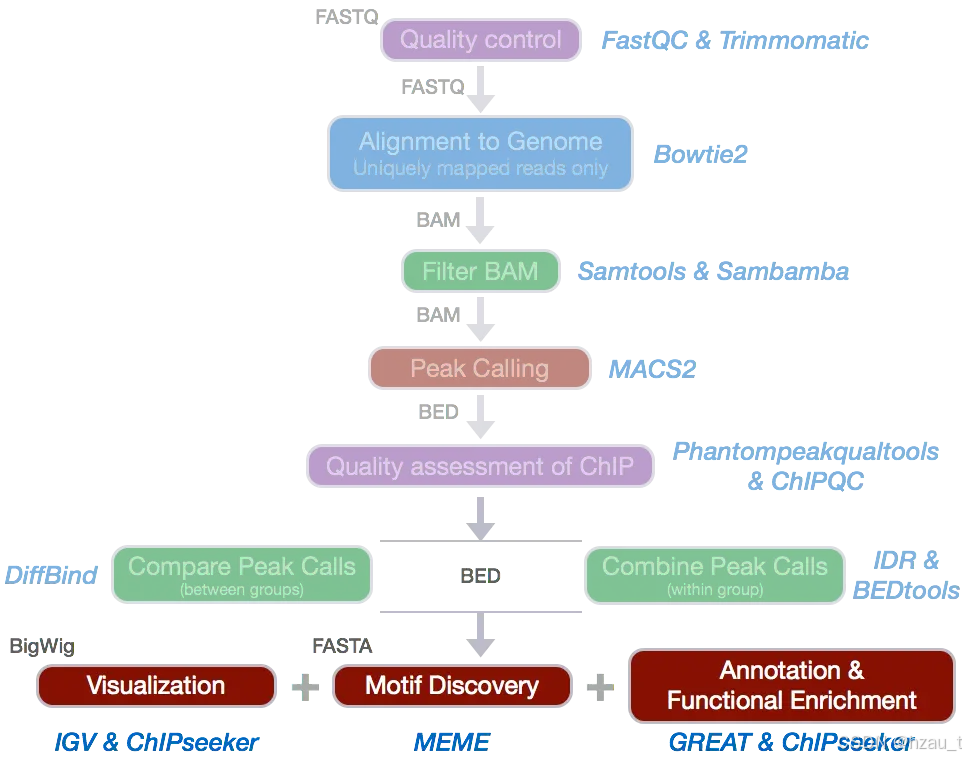
1 环境安装及配置
conda create -n atac
source activate atac
#安装anaconda后,需要配置清华镜像源
conda install -c conda-forge bowtie2 fastqc trim_galore deeptools sambamba samtools 2 raw_data QC
#fastqc对所有rawdata进行数据质控
fastqc -t 30 /home/ruiqing/rawdata/*gz -o rawqc/3 数据过滤、去接头
#在rawdata下构建,然而rawdata文件夹由root账号创建,只读
#创建/home/ruiqing/config文件夹/构建config文件
ls *1.fq.gz > 1
ls *2.fq.gz > 2
paste 1 1 2 > config
vim config
#修改第一列的名称,调整为列名
#用于trim.sh
#用于trim接头和低质量序列
cat raw.config |while read id;
do echo $id
arr=($id)
fq2=${arr[2]}
fq1=${arr[1]}
sample=${arr[0]}
echo $sample
trim_galore -q 25 --phred33 --length 35 -e 0.1 --stringency 4 --paired -o ../clean ../rawdata/$fq1 ../rawdata/$fq2
done4 bowtie2 比对至参考基因组上,得到raw.bam文件
#在clean下构建
ls *1.fq.gz > 1
ls *2.fq.gz > 2
paste 1 1 2 > config
vim config
#修改第一列的名称,调整为列名#bowtie2.sh
cat config|while read id;
do echo $id
arr=($id)
fq1=${arr[1]}
fq2=${arr[2]}
sample=${arr[0]}
bowtie2 -p 20 --very-sensitive -X 2000 -x /home/ruiqing/reference/ricegenome -1 $fq1 -2 $fq2 | samtools sort -T ./ -O bam -@ 20 -o - > /home/ruiqing/align/${sample}.bam
samtools index /home/ruiqing/align/${sample}.bam
samtools flagstat /home/ruiqing/align/${sample}.bam > /home/ruiqing/align/${sample}.stat
done
#flagstat需要写绝对路径比对结果,原始bowtie.sh文件中flagstat没有写绝对路径,导致没有输出stat文件并报错
5 对raw.bam文件去重、过滤
mkdir -p /home/ruiqing/rmdup
mkdir -p /home/ruiqing/lastbamcat /home/ruiqing/clean/config|while read id;
do echo $id
arr=($id)
fq1=${arr[1]}
fq2=${arr[2]}
sample=${arr[0]}
sambamba markdup --overflow-list-size 600000 -t 20 --tmpdir='./' -r /home/ruiqing/align/${sample}.bam /home/ruiqing/rmdup/${sample}.rmdup.bam
samtools index /home/ruiqing/rmdup/${sample}.rmdup.bam
samtools flagstat /home/ruiqing/rmdup/${sample}.rmdup.bam > /home/ruiqing/rmdup/${sample}.rmdup.stat
samtools view -h -f 2 -q 30 /home/ruiqing/rmdup/${sample}.rmdup.bam |grep -v ChrUn|grep -v ChrSy|samtools sort -T ./ -O bam -@ 20 -o - 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 1577
1577

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









