




 本文介绍了GFF3和GTF两种基因组注释格式,包括它们的结构、用途以及如何使用gffread进行格式转换。GFF3用于存储序列结构信息,每个条目包含9个属性,如seqid、start、end等。GTF主要用于基因注释,其第9列必须包含gene_id和transcript_id。此外,文章还提到了利用bedtools和jcvi库提取序列以及获取最长转录本的方法。
本文介绍了GFF3和GTF两种基因组注释格式,包括它们的结构、用途以及如何使用gffread进行格式转换。GFF3用于存储序列结构信息,每个条目包含9个属性,如seqid、start、end等。GTF主要用于基因注释,其第9列必须包含gene_id和transcript_id。此外,文章还提到了利用bedtools和jcvi库提取序列以及获取最长转录本的方法。
GFF3 官方
- General Feature Format Version 3
- 存储
序列结构信息的一种数据格式。序列结构就是一个scaffold或者染色体上面每个位置都是什么序列元件。 - GFF每一行代表一个序列元件(以#为开头的注释行除外),一行9列9个属性,必须tab分割,属性为空用“.”代替。
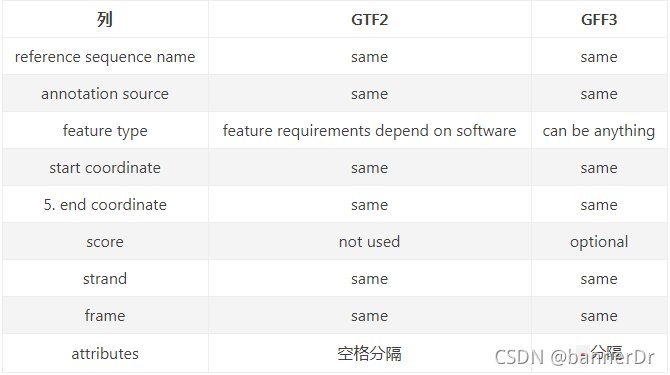
1. seqid - scaffold或者chromosome的名称说明
2. source - 产生一个序列元件的软件的名称或者数据源(数据库名称或者项目名称)
3. type - 序列元件的类型,例如:mRNA、CDS等等
4. start - 序列元件在scaffold或者chromosome上的起始位置,从1开始计数
5. end - 序列元件在scaffold或者chromosome上面的终止位置,从1开始计数
6. score - 该序列元件的打分,一般为该序列元件做比对时的E-value和ab initio gene prediction features时的P-value
7. strand - “+”代表该序列元件在scaffold或者chromosome的正链,反之亦反
8. phase - 可以为“0”、“1”、“2”,“0”代表该序列元件的第一个碱基为第一个密码子的第一个剪辑,“1”代表该序列元件的第二个碱基是第一个密码子 的第一个碱基,依次类推。
9. attributes - 该序列元件的一些其他属性,可以有多个每个属性之间必须以“;”分割,例如“ID=some-id;Name=some-name;Parent=some-parent”,请注意这个Parent属性,由于序列元件是很复杂的,一个序列元件(例如:exon)可能属于另外一个序列元件(例如:gene),这个Parent属性的意思就是该序列元件在哪个序列元件上面,如果一个序列元件没有Parent属性,说明他的父元件就是scaffold或者chromosome
GFF文件的第9列,从第二版开始(GFF2),所有的属性都以标签=值的方式呈现,各个属性之间以;作为分隔符
GTF官方
当前所广泛使用的GTF格式为第二版(GTF2),它主要是用来描述基因的注释。GTF格式有两个硬标准:
- 根据所使用的软件的不同,feature types是必须注明的。
- 第9列必须以gene_id以及transcript_id开头。GTF文件的第9列同GFF文件不同,虽然同样是标签与值配对的情况,但标签与值之间以空格分开,且每个特征之后都要有分号;(包括最后一个特征):
格式转换
Cufflinks里面的工具gffread
#gff2gtf
gffread my.gff3 -T -o my.gtf
#gtf2gff
gffread merged.gtf -o- > merged.gff3
gffread genome.gff3 -g genome.fa -x cds.fa
gffread genome.gff3 -g genome.fa -y protein.fa
gffread genome.gff3 -g genome.fa -w transcripts.fa
python -m jcvi.formats.gff bed --type=mRNA --key=ID a.gff3 > mRNA.bed
#Python 包jcvi包, 抽取 GFF文件中mRNA, key是ID
#基因
python -m jcvi.formats.gff bed --type=gene --key=ID a.chr.gff3 > gene.bed
用bedtools提取序列,要加-s 参数(区分正反链)
bedtools getfasta -fi ./a.fasta -bed gene.bed -nameOnly -s -fo a.gene.fa
a.gene.fa就是我们需要的文件,同理也可以得到a.mRNA.fa。
提取最长转录本脚本,获得基因中最长可变剪切的序列。

















 583
583

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









