







单细胞转录组 —— STARsolo 原始数据处理实战
前言
前面我们已经介绍了几种原始数据处理工具,最后再介绍一种多平台兼容的快速定量工具 —— STARsolo。
主要使用的还是 STAR 比对软件,只是增加了更多对单细胞数据的处理,不同平台数据的差异,也只是在参数设置上。
实战
软件可以直接用 conda 进行安装
conda create -n STAR -c bioconda -c conda-forge star seqtk
下载人类参考基因组
wget http://ftp.ensembl.org/pub/release-98/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
wget http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.primary_assembly.annotation.gtf.gz
或者小鼠参考基因组
wget ftp://ftp.ensembl.org/pub/release-98/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.primary_assembly.fa.gz
wget ftp://ftp.ensembl.org/pub/release-98/gtf/mus_musculus/Mus_musculus.GRCm38.98.gtf.gz
解压文件
gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
gunzip gencode.v32.primary_assembly.annotation.gtf.gz
gunzip Mus_musculus.GRCm38.dna.primary_assembly.fa.gz
gunzip Mus_musculus.GRCm38.98.gtf.gz
构建索引
构建人类参考基因组索引
STAR \
--runThreadN 20 \
--runMode genomeGenerate \
# 索引保存路径
--genomeDir human \
# fa 文件路径
--genomeFastaFiles Homo_sapiens.GRCh38.dna.primary_assembly.fa \
# gtf文件路径
--sjdbGTFfile gencode.v32.primary_assembly.annotation.gtf
类似地,可以构建小鼠参考基因组索引
STAR \
--runThreadN 20 \
--runMode genomeGenerate \
# 索引保存路径
--genomeDir mmu \
# fa 文件路径
--genomeFastaFiles Mus_musculus.GRCm38.dna.primary_assembly.fa \
# gtf文件路径
--sjdbGTFfile Mus_musculus.GRCm38.98.gtf
scRNA-seq
从 SRA 数据库中下载 PRJNA666791 项目的 10x scRNA-seq 数据进行分析
prefetch --option-file SRR_Acc_List.txt --output-directory sra
解压数据
ls sra/*/*.sra | xargs fastq-dump --split-files --gzip -O raw
10x barcode 文件可以在下载的 cellranger 软件中找到,例如
ls /path/to/cellranger-8.0.0/lib/python/cellranger/barcodes
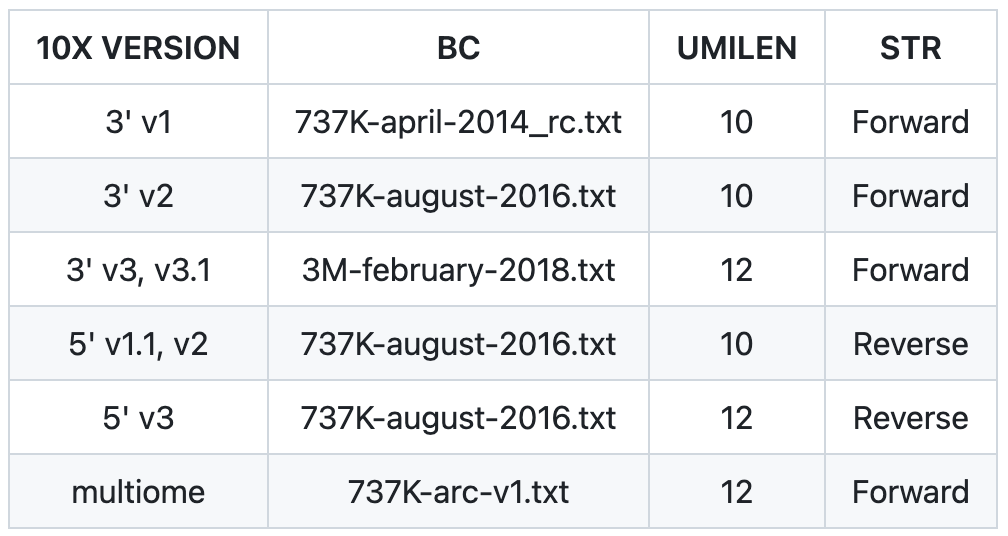
参数配置,需要根据所使用数据的信息进行调整
```bash
# 白名单
BC=737K-august-2016.txt
# barcode 的长度
CBLEN=16
# UMI 的长度
UMILEN=10
# 文库方向
STRAND=Forward # Forward/Reverse/Unstranded
FQDIR="$(pwd)/raw"
TAG="GSM4812353"
R1=""
R2=""
if [[ `find  最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 1716
1716

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











