



 作者在使用STAR-solo进行NCBI上的smartseq数据集定量后,遇到使用scanpy的sc.read_10x_mtx函数读取文件时的ValueError。问题源于matrix.mtx文件中的行索引超过矩阵维度。通过检查文件发现一行基因编号异常,修正后解决了问题。
作者在使用STAR-solo进行NCBI上的smartseq数据集定量后,遇到使用scanpy的sc.read_10x_mtx函数读取文件时的ValueError。问题源于matrix.mtx文件中的行索引超过矩阵维度。通过检查文件发现一行基因编号异常,修正后解决了问题。
由于演示需要,今天用STAR-solo给NCBI上随便找的一个smartseq数据集做了定量(PRJNA591860),在定量之后,得到了常规的三个文件,压缩后下载到本地,到这里一切正常。(BTW,用solo做smartseq定量真的好方便,不用自己合并了)
在使用scanpy的sc.read_10x_mtx读取上述文件时,幺蛾子出现了。
---------------------------------------------------------------------------
ValueError Traceback (most recent call last)
Cell In[65], line 1
----> 1 adata = sc.read_10x_mtx("./raw/", var_names='gene_symbols')
File D:\Anaconda\envs\sc10\lib\site-packages\scanpy\readwrite.py:490, in read_10x_mtx(path, var_names, make_unique, cache, cache_compression, gex_only, prefix)
488 genefile_exists = (path / f'{prefix}genes.tsv').is_file()
489 read = _read_legacy_10x_mtx if genefile_exists else _read_v3_10x_mtx
--> 490 adata = read(
491 str(path),
492 var_names=var_names,
493 make_unique=make_unique,
494 cache=cache,
495 cache_compression=cache_compression,
496 prefix=prefix,
497 )
498 if genefile_exists or not gex_only:
499 return adata
File D:\Anaconda\envs\sc10\lib\site-packages\scanpy\readwrite.py:554, in _read_v3_10x_mtx(path, var_names, make_unique, cache, cache_compression, prefix)
550 """
551 Read mtx from output from Cell Ranger v3 or later versions
552 """
553 path = Path(path)
--> 554 adata = read(
555 path / f'{prefix}matrix.mtx.gz',
556 cache=cache,
557 cache_compression=cache_compression,
558 ).T # transpose the data
559 genes = pd.read_csv(path / f'{prefix}features.tsv.gz', header=None, sep='\t')
560 if var_names == 'gene_symbols':
File D:\Anaconda\envs\sc10\lib\site-packages\scanpy\readwrite.py:112, in read(filename, backed, sheet, ext, delimiter, first_column_names, backup_url, cache, cache_compression, **kwargs)
110 filename = Path(filename) # allow passing strings
111 if is_valid_filename(filename):
--> 112 return _read(
113 filename,
114 backed=backed,
115 sheet=sheet,
116 ext=ext,
117 delimiter=delimiter,
118 first_column_names=first_column_names,
119 backup_url=backup_url,
120 cache=cache,
121 cache_compression=cache_compression,
122 **kwargs,
123 )
124 # generate filename and read to dict
125 filekey = str(filename)
File D:\Anaconda\envs\sc10\lib\site-packages\scanpy\readwrite.py:751, in _read(filename, backed, sheet, ext, delimiter, first_column_names, backup_url, cache, cache_compression, suppress_cache_warning, **kwargs)
749 adata = read_excel(filename, sheet)
750 elif ext in {'mtx', 'mtx.gz'}:
--> 751 adata = read_mtx(filename)
752 elif ext == 'csv':
753 adata = read_csv(filename, first_column_names=first_column_names)
File D:\Anaconda\envs\sc10\lib\site-packages\anndata\_io\read.py:316, in read_mtx(filename, dtype)
313 from scipy.io import mmread
315 # could be rewritten accounting for dtype to be more performant
--> 316 X = mmread(fspath(filename)).astype(dtype)
317 from scipy.sparse import csr_matrix
319 X = csr_matrix(X)
File D:\Anaconda\envs\sc10\lib\site-packages\scipy\io\_mmio.py:129, in mmread(source)
84 def mmread(source):
85 """
86 Reads the contents of a Matrix Market file-like 'source' into a matrix.
87
(...)
127 [0., 0., 0., 0., 0.]])
128 """
--> 129 return MMFile().read(source)
File D:\Anaconda\envs\sc10\lib\site-packages\scipy\io\_mmio.py:579, in MMFile.read(self, source)
577 try:
578 self._parse_header(stream)
--> 579 return self._parse_body(stream)
581 finally:
582 if close_it:
File D:\Anaconda\envs\sc10\lib\site-packages\scipy\io\_mmio.py:814, in MMFile._parse_body(self, stream)
810 od_V = od_V.conjugate()
812 V = concatenate((V, od_V))
--> 814 a = coo_matrix((V, (I, J)), shape=(rows, cols), dtype=dtype)
815 else:
816 raise NotImplementedError(format)
File D:\Anaconda\envs\sc10\lib\site-packages\scipy\sparse\_coo.py:197, in coo_matrix.__init__(self, arg1, shape, dtype, copy)
194 if dtype is not None:
195 self.data = self.data.astype(dtype, copy=False)
--> 197 self._check()
File D:\Anaconda\envs\sc10\lib\site-packages\scipy\sparse\_coo.py:284, in coo_matrix._check(self)
282 if self.nnz > 0:
283 if self.row.max() >= self.shape[0]:
--> 284 raise ValueError('row index exceeds matrix dimensions')
285 if self.col.max() >= self.shape[1]:
286 raise ValueError('column index exceeds matrix dimensions')
ValueError: row index exceeds matrix dimensions
很容易确认这个报错来自于matrix.mtx文件(因为另外两个文件也不可能出什么问题)
单独使用sc.read_10x_mtx和scipy.io.mmread读取该mtx文件,仍然出现了和上述一样的报错,确定了报错源就是这个mtx文件的row index exceeds matrix dimensions。
但这句话到底说明了什么呢。完全不能理解。我尝试了一下午,从mtx的行数检查到文末空格是否添加,以及重新做了一遍star的reference,这个报错就是纹丝不动。谷歌上也没有找到和我有同样遭遇的人。
最后在经过三个多小时的排查之后终于发现了问题的源头。
使用with open的方式在python里打开这个mtx文件,逐行检查每一行的第一个数字是不是小于36601(定量之后的基因总数)
count = 0
with open("./result/matrix.mtx", 'r') as f:
for line in f.readlines():
count += 1
try:
int(line.split(" ")[0])
except:
continue
if int(line.split(" ")[0])>36601:
print(count, line)
然后发现:

??总共才36601个基因,居然在这一行的第一个数字上出现了一个一百多万。
直接在txt里删除这行,然后在表头位置的总行数(57328)上减了1,重新读取。
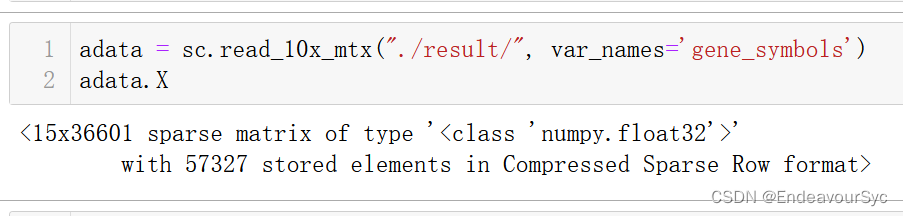
我真是日了狗了......到现在我还是不清楚为什么定量完的结果会在基因编号这种地方出错误,还错的这么离谱 。说来也是倒霉,这个数据集一共2w多个样本,随便划了15个就碰到了这种问题。

最后复习一下单细胞定量之后的mtx文件的含义,如上图,前两行是稀疏矩阵的固定格式,第三行中,前两个数字(36601和15)分别表示这一列里出现的数字的上限,最后一个57328表示这个文件的总行数。之后的每一行(x,y,z)分别表示在稀疏矩阵的x行y列处有值z。
这个美好的下午就被这个离谱的报错毁了...

















 3139
3139

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









