






乡亲们,祝大家新的元年财源广进,幸福和谐。今天跟大家分享,如何利用R RcisTarget 包预测基因转录因子。
1.R RcisTarget 获取
使用 Bioconductor 安装
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("RcisTarget")
或者直接在官网下载,本地安装,此处不再赘述。
2.其它配置文件
此处我们还需结合基因座注释文件,一般是TSS+/-10kb范围的,可在:Homo sapiens
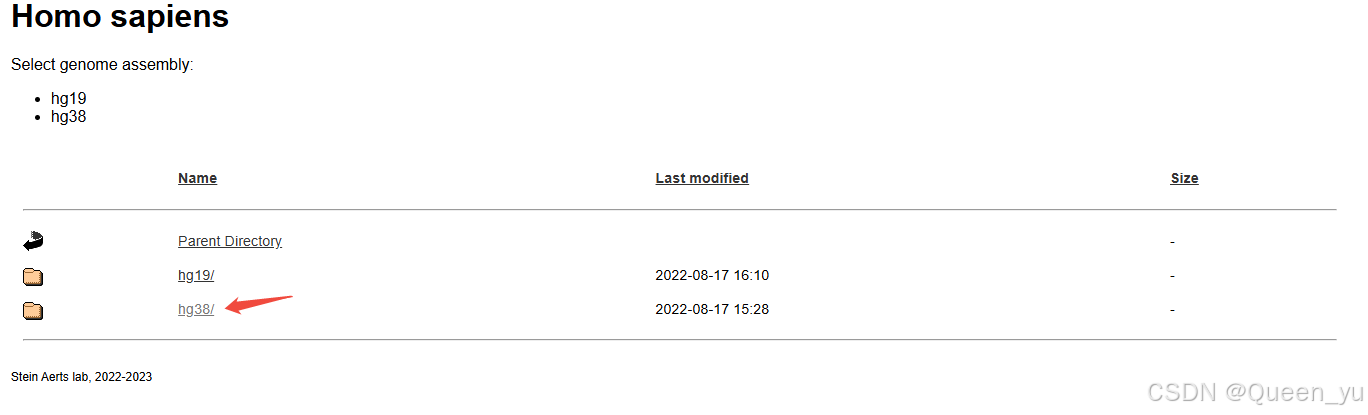
获取所需配置文件:
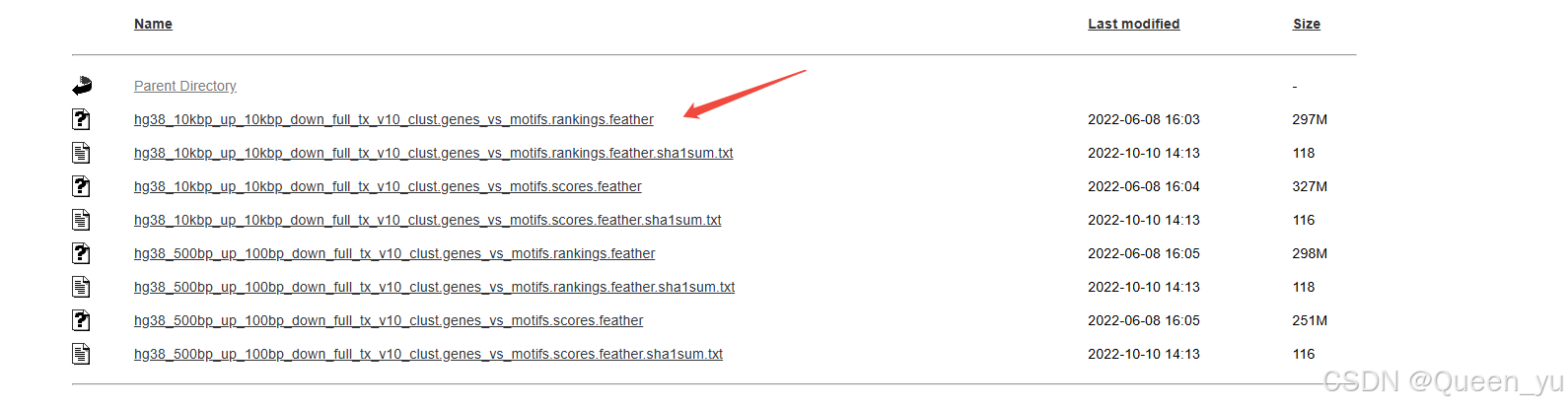
3.转录因子预测
通过以上步骤,我们把转录因子预测所需环境搞定,接下来开始正式预测,博主这里是核心基因--通过生物信息学手段如机器学习和实验验证获得的基因--xxx和xx,即一个上调和一个下调基因,基本是符合该包最低的数据输入格式类型。
rm(list = ls()) #清空当前环境
library(RcisTarget) #调用包
setwd("E:/05.F20240902021/25.TF") #设置分析环境
# 加载数据
data(motifAnnotations_hgnc) #加载包内置文件
motifRankings <- importRankings("hg38_10kbp_up_10kbp_down_full_tx_v10_clust.genes_vs_motifs.rankings.feather") #预测范围
# 创建包含目标基因的基因集
genelists <- list("upgenes" = 'xxx', "downgenes" = 'xx') # 结合目标基因在数据中具体情况
# 使用cisTarget函数进行转录因子预测
motifEnrichmentTable_wGenes <- cisTarget(
geneSets = genelists,
motifRankings = motifRankings,
motifAnnot = motifAnnotations,
highlightTFs = NULL,
nesThreshold = 3,
geneErnMethod = "iCisTarget",
geneErnMaxRank = 5000,
nCores = 4,
verbose = TRUE
)
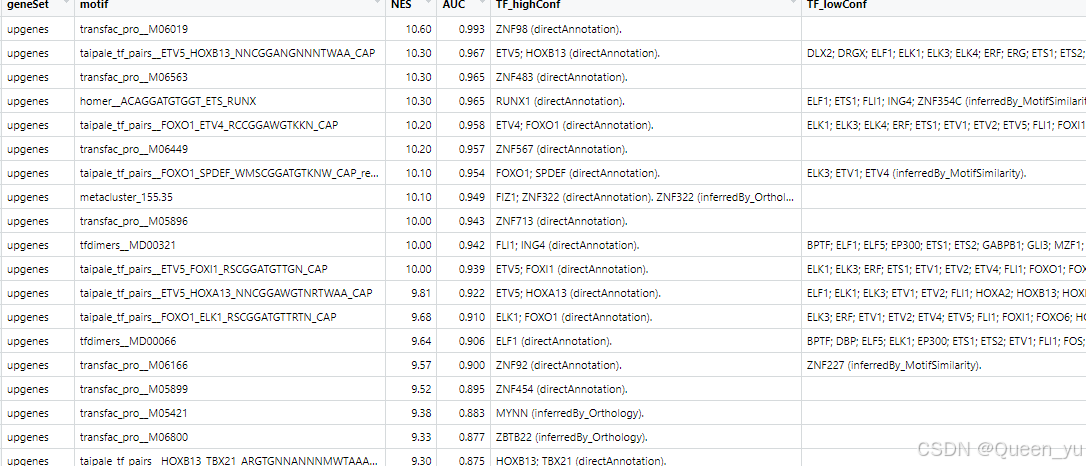
跑完上边代码,会出现的结果其中:
1.geneSet: 分析的基因集合。这里显示为 upgenes,表示这些 motif 是在上调基因中富集的结果。
2.motif:表示该行数据对应的转录因子结合位点(motif)。它可能来源于某个 motif 数据库(如 JASPAR 或 HOMER)。
3.NES: Normalized Enrichment Score,标准化富集评分。该值表示 motif 在基因集合中的富集程度,数值越高说明富集越显著。一般来说,NES > 3 被认为是显著富集。
4.AUC: Area Under the Curve,富集分析的曲线下面积。表示 motif 和目标基因集合之间的关系强度,范围通常在 [0, 1],数值越大说明关联性越强。
5.TF_highConf/lowcont: Motif 对应的高置信度(低)转录因子(TF)。
后续博主重点关注NES值和AUC值---AUC > 0.8 。
#输出结果
write.table(motifEnrichmentTable_wGenes,
file = "transcription_factors_filtered.txt",
sep = "\t",
quote = FALSE,
row.names = FALSE)


















 9201
9201

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









