



简单介绍
ZDOCK:ZDOCK是一种用于预测蛋白质-蛋白质互作(PPI)的复合物结构的程序。它使用了一个基于快速傅立叶转换(FFT)的搜索算法,允许全面探索蛋白质之间可能的结合方式。ZDOCK的评分函数考虑了分子间的静电相互作用,范德华力,和解离自由能等。然后,它生成一个打分排名的预测复合物列表。ZDOCK可以用于抗体-抗原,酶-底物,蛋白质-核酸等各种类型的PPI预测。
ZRANK:ZRANK是一个独立的打分函数,用于对ZDOCK预测的蛋白质复合物进行再排序。这个函数包括了范德华力,静电相互作用,以及解离自由能。ZRANK打分通常可以提高ZDOCK的预测精度,因此它们经常一起使用。
RDOCK:RDOCK是一个结构基础的虚拟筛选软件,用于预测小分子配体和蛋白质的结合模式和结合能。RDOCK是DOCK的一部分,它利用蛋白质的已知3D结构,通过分子对接的方法来预测小分子的最佳结合模式。
下载链接
蛋白质对接软件(ZDOCK & RDOCK) --- Protein Docking Software (ZDOCK & RDOCK)
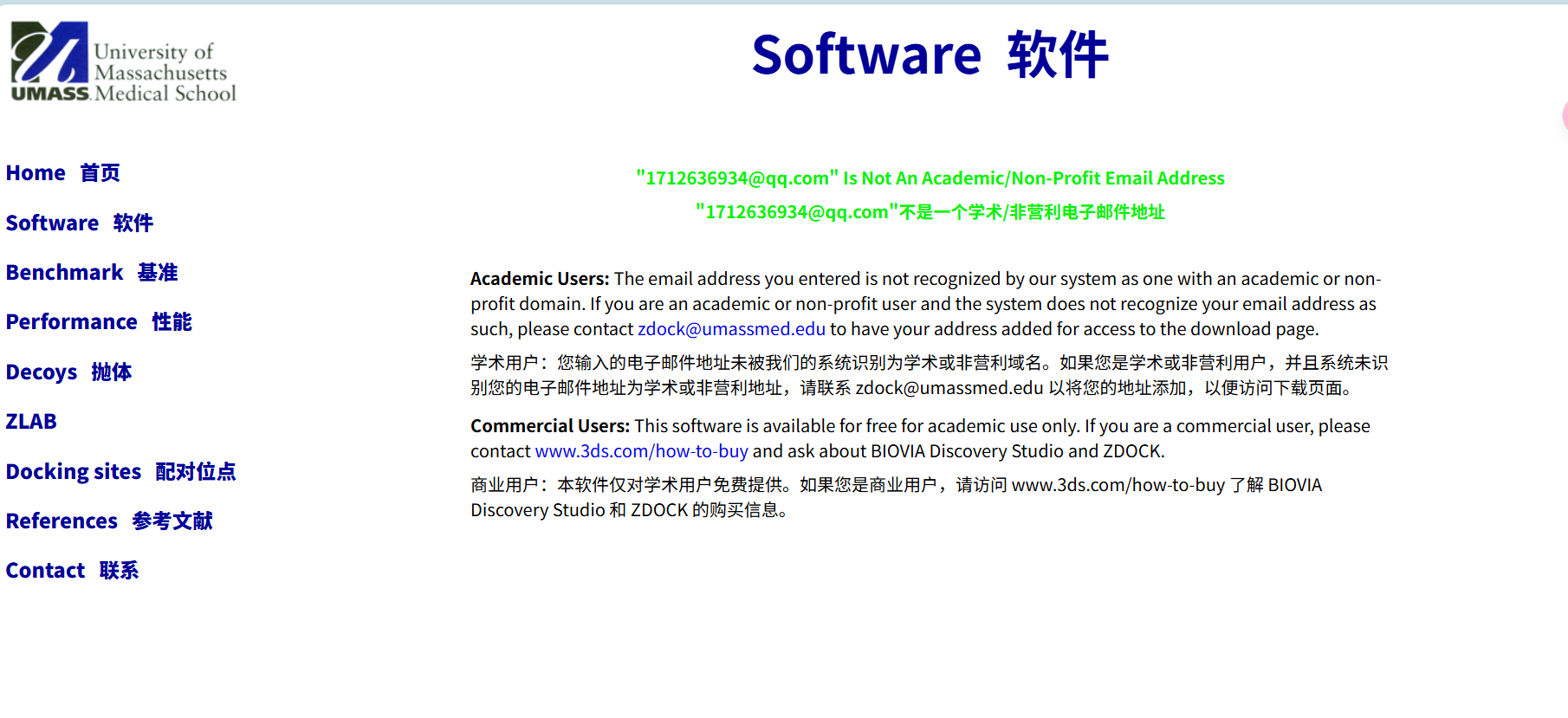
未注册?请输入学术或非营利性电子邮件地址,我们将发送一个有效期为 24 小时的密码。
ZDOCK、M-ZDOCK、ZRANK 和 RDOCK 可通过 BIOVIA 商业获得。
| If you are a commercial user, please go to www.3ds.com/how-to-buy and ask about BIOVIA Discovery Studio and ZDOCK. |
发邮件
Hello,
We are a group of academic researchers dedicated to advancing the field of molecular biology, and we hope to utilize the zdock software as part of our efforts to predict protein-protein interactions (PPI). Protein-protein interactions play a crucial role in many biological processes, including signal transduction, immune response, and cellular metabolism. By understanding these interactions, we aim to contribute to the broader scientific community's knowledge of how proteins function together within complex biological systems.
Zdock is renowned for its ability to provide accurate docking predictions, which can significantly enhance our research capabilities. The software employs advanced algorithms that allow for efficient sampling of possible protein orientations and evaluation of their binding affinities. This capability is particularly valuable for studying large-scale PPI networks or exploring potential therapeutic targets where experimental methods may be limited or time-consuming.
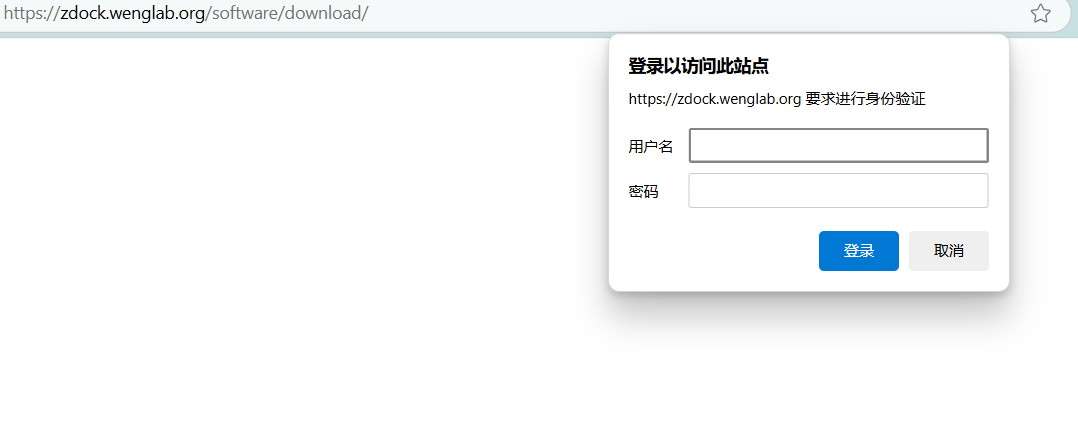
To facilitate our research and ensure compliance with the licensing terms for academic use, we kindly request that you add our email address to the approved academic user list. This will enable us to access the download page and integrate zdock into our computational workflows. We assure you that the software will be used exclusively for non-commercial, academic purposes aimed at furthering scientific discovery and education.
By supporting initiatives such as ours, you are also contributing to the ongoing development of biology as a discipline. Our findings could lead to new insights into disease mechanisms, drug design strategies, or fundamental cellular processes. Therefore, your assistance in granting us access to zdock would be greatly appreciated.
Thank you for considering our request. Please let us know if any additional information is required to expedite this process. We look forward to collaborating with the zdock team and leveraging this powerful tool in our pursuit of scientific advancement.
Best regards,
[Your Name/Organization]
你好,
我们是一群致力于推动分子生物学领域发展的学术研究人员,希望利用 zdock 软件作为我们预测蛋白质 - 蛋白质相互作用(PPI)工作的一部分。蛋白质 - 蛋白质相互作用在许多生物过程中起着关键作用,包括信号转导、免疫反应和细胞代谢。通过了解这些相互作用,我们旨在为更广泛的科学界对复杂生物系统中蛋白质如何协同工作的认识做出贡献。
Zdock 以其能够提供精准的对接预测而闻名,这能显著提升我们的研究能力。该软件采用先进的算法,能够高效地对可能的蛋白质取向进行采样,并评估其结合亲和力。这种能力对于研究大规模的蛋白质相互作用网络或探索潜在的治疗靶点尤其有价值,在这些情况下,实验方法可能受到限制或耗时过长。
为了便于我们的研究并确保符合学术用途的许可条款,我们恳请您将我们的电子邮箱地址添加到获批的学术用户列表中。这将使我们能够访问下载页面,并将 zdock 集成到我们的计算工作流程中。我们向您保证,该软件将仅用于非商业性的学术目的,旨在促进科学发现和教育。
通过支持像我们这样的倡议,您也在为生物学这一学科的持续发展做出贡献。我们的研究结果可能会为疾病机制、药物设计策略或基本细胞过程带来新的见解。因此,若您能协助我们使用 zdock,我们将不胜感激。
感谢您考虑我们的请求。如有任何需要额外提供的信息以加快这一进程,请告知我们。我们期待与 zdock 团队合作,并利用这一强大的工具来推进我们的科学研究。
致以最诚挚的问候填写非盈利邮箱账号
报错及解决办法
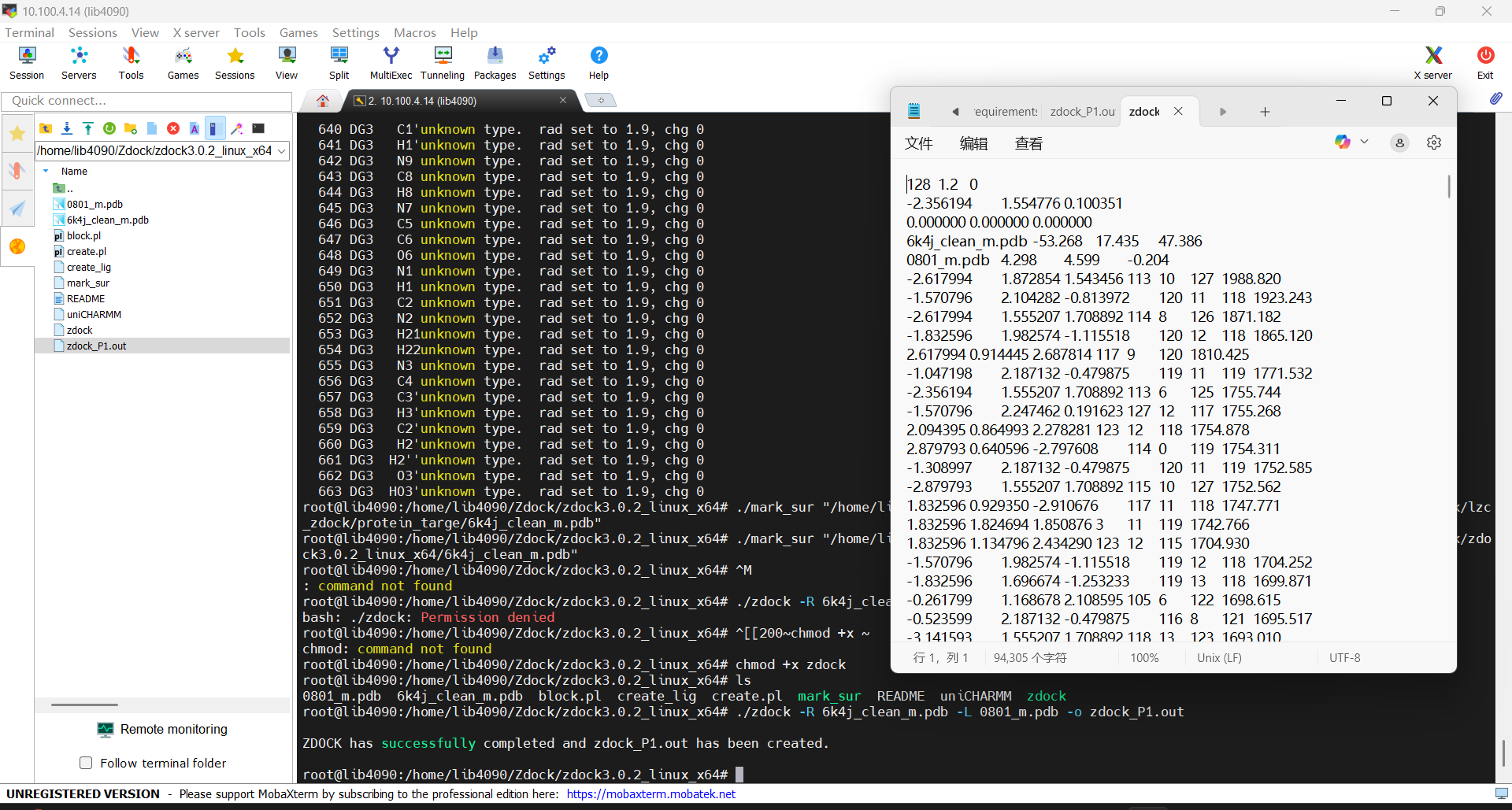
设置文件为可执行
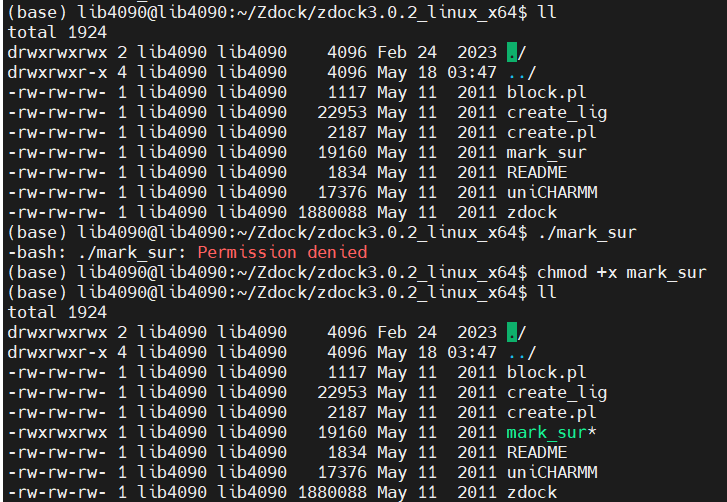
要使文件具有执行权限,可以使用chmod命令。以下是一些常用的命令示例:
# 使文件对所有用户可执行
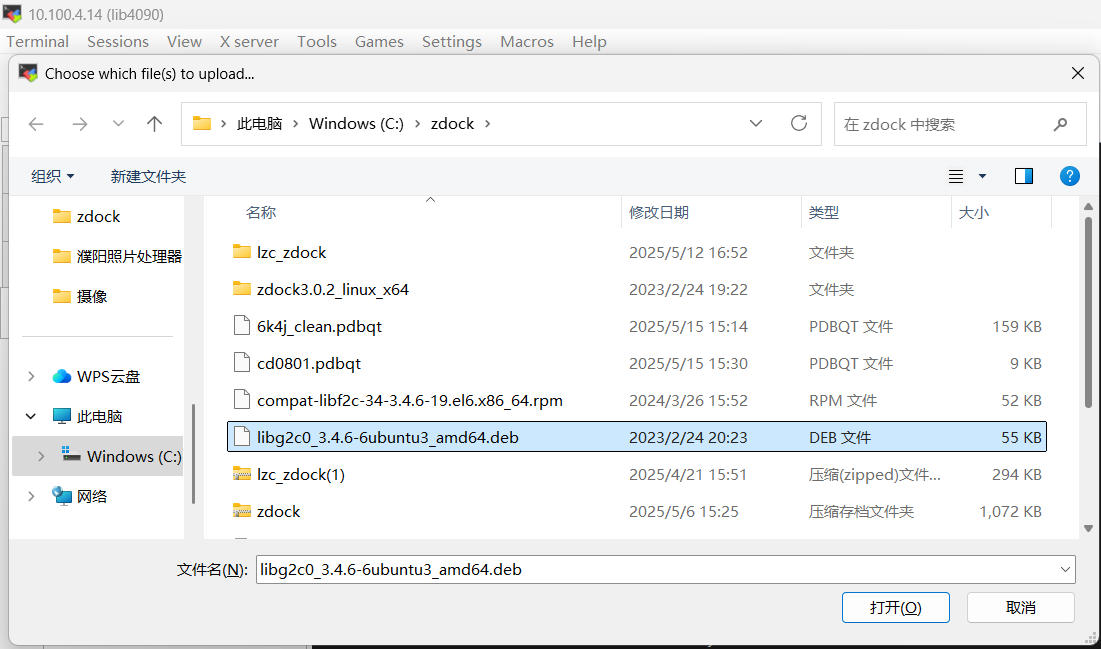
chmod +x 文件名/group_share/zdock/zdock/extract/usr/lib
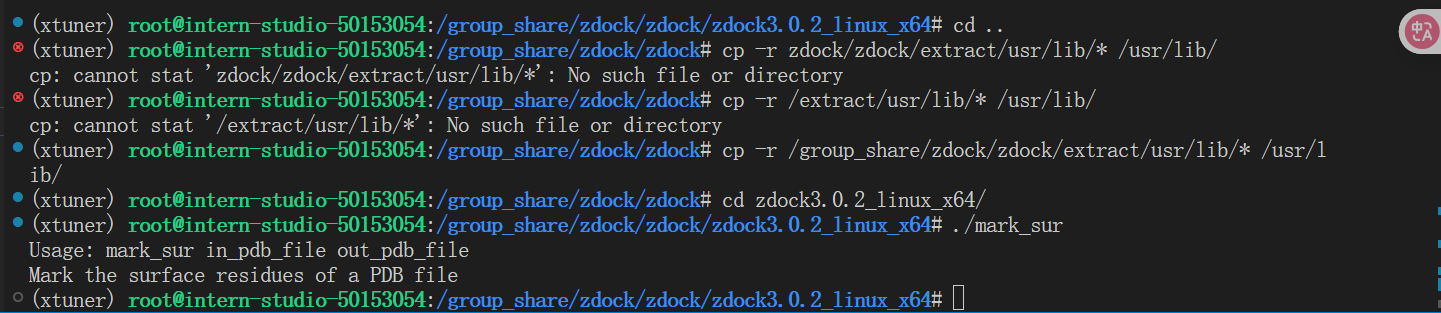
cp -r /ex/* /usr/lib/
解压 deb 包
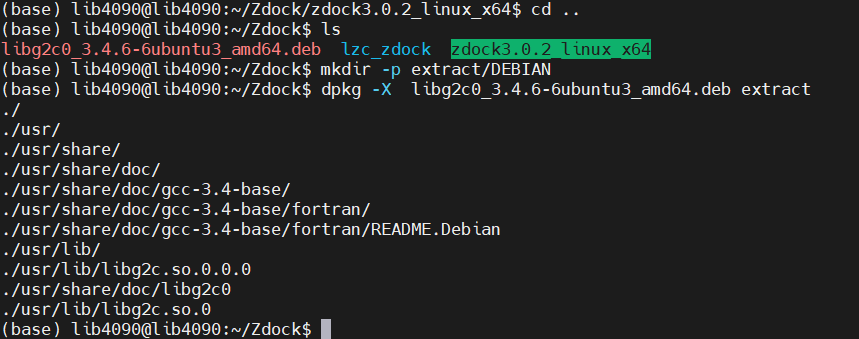
在 Ubuntu 或 Debian 系统上,解压 .deb 包文件有两种常见方法:使用 dpkg 命令或 ar 命令。
使用 dpkg 命令
创建一个目录来存放解压后的文件: mkdir -p extract/DEBIAN
使用 dpkg 命令解压 .deb 包: dpkg -X ./package.deb extract
解压 .deb 包中的控制信息: dpkg -e ./package.deb extract/DEBIAN
mkdir -p extract/DEBIAN
dpkg -X libg2c0_3.4.6-6ubuntu3_amd64.deb extract
dpkg -e libg2c0_3.4.6-6ubuntu3_amd64.deb extract/DEBIAN
cp -r /home/lib4090/Zdock/extract/usr/lib/* /usr/lib/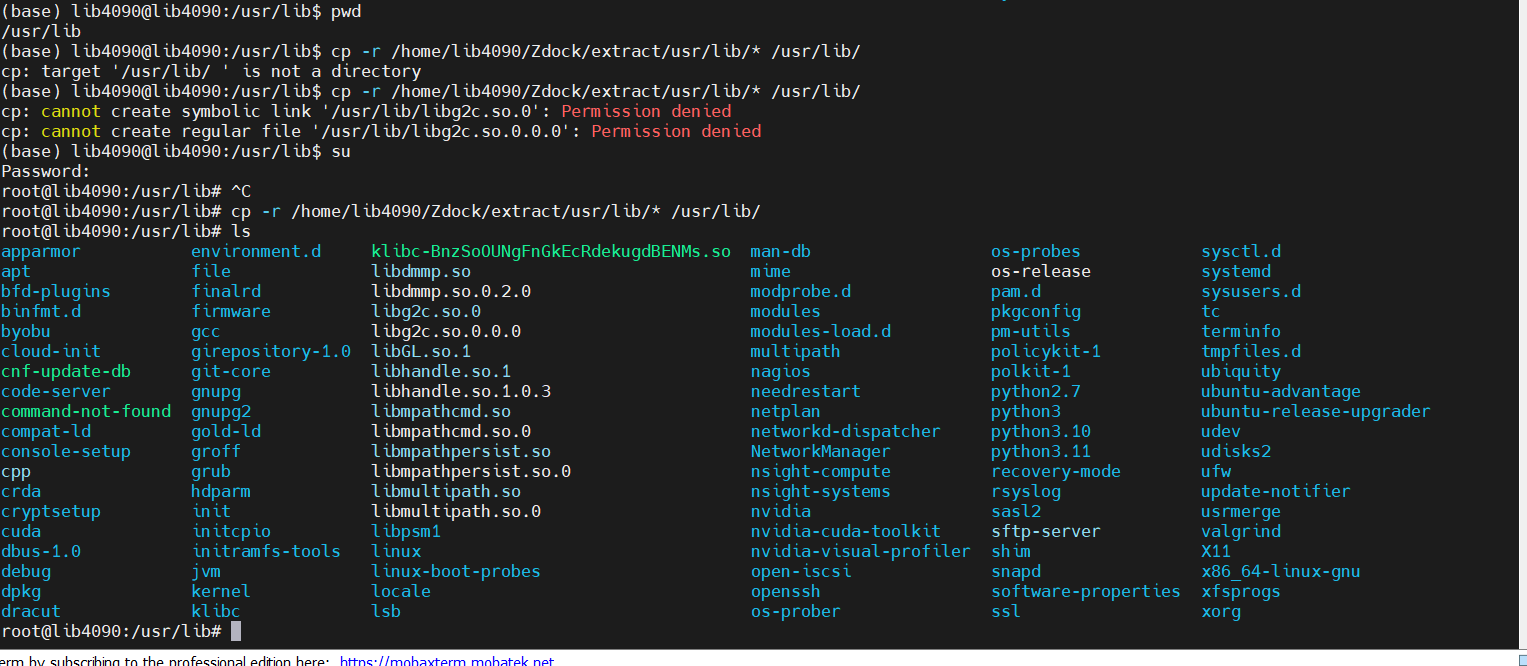
Usage
Usage: zdock [options] -R [receptor.pdb] -L [ligand.pdb]
Standard PDB format files must be processed by mark_sur to add atom radius, charge and atom type. If you know that some atoms are not in the binding site, you can block them by changing their atom type (column 55-56) to 19.
Available options:
-o filename : set the output filename (default to zdock.out)
-N int : set the number of output predictions, (default to 2000)
-S int : set the seed for the randomization of the starting PDBs (default to no randomization)
-D : set rotational sampling to be dense, used only if zdock output will be further processed by more detailed binding energy calculations (default to none)
-F : fix the receptor, preventing its rotation and switching with ligand during execution.Example:
mark_sur receptor.pdb receptor_m.pdb
mark_sur ligand.pdb ligand_m.pdb
zdock -R receptor_m.pdb -L ligand_m.pdb -o zdock.out
create.pl zdock.out
#以下是使用 Matplotlib 绘制类似图表的代码,已优化为方便复制的格式:
import matplotlib.pyplot as plt
import numpy as np
methods = {'zDcK': {'P1': 1894.783, 'P2': 2154.751,'P3': 2024.333,'P4': 1988.820,'CD63': 0000.000}.'AptaTrans': {'P1': 0.91850644,'P2': 0.7680103,'P3': 0.5980261,'P4': 0.00893546,'CD63': 0.00833475}}
targets ='P1','P2','P3','P4','CD63']
plt.figure(figsize=(12,8))
bar_width = 0.25
index = np.arange(len(targets))
colors =['limegreen',"cornflowerblue','darkgrey']
for i,(method, color)in enumerate(zip(methods.keys(), colors)):plt,barh(index + i * bar_width, [methods[method][t] for t in targets], bar_width, label=method, color=color)for j,target in enumerate(targets):
plt.text(methods[methodl[target] + 10, i + i * bar width, f"fmethods[methodl[targetl:.1f}", va='center', color='black')
plt.xlabel('ZD0CK/AptaTrans Score',fontsize=12)
plt.ylabel('Aptamer',fontsize=12)
plt.title('Performance Comparison for Aptamer Recommendation', fontsize=14)
plt.yticks(index + bar width, targets, fontsize=10)
plt.legend(loc='upper right',fontsize=10)
plt.grid(axis='x',linestyle='--',alpha=0.7)
plt.tight layout()
plt.show() 
进入
本文参考:————————————————————————————————---------————
linux下zdock与zrank安装及基础使用方法 - Kngiht_of_Night的个人博客
Ubuntu系统下deb包的解压、打包、安装、卸载及常用命令_ubuntu解压deb-优快云博客
zdock批量对接 python

















 3503
3503

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









