



LightDock: a new multi-scale approach to protein–protein docking
The LightDock server is free and open to all users and there is no login requirement
server
1示例
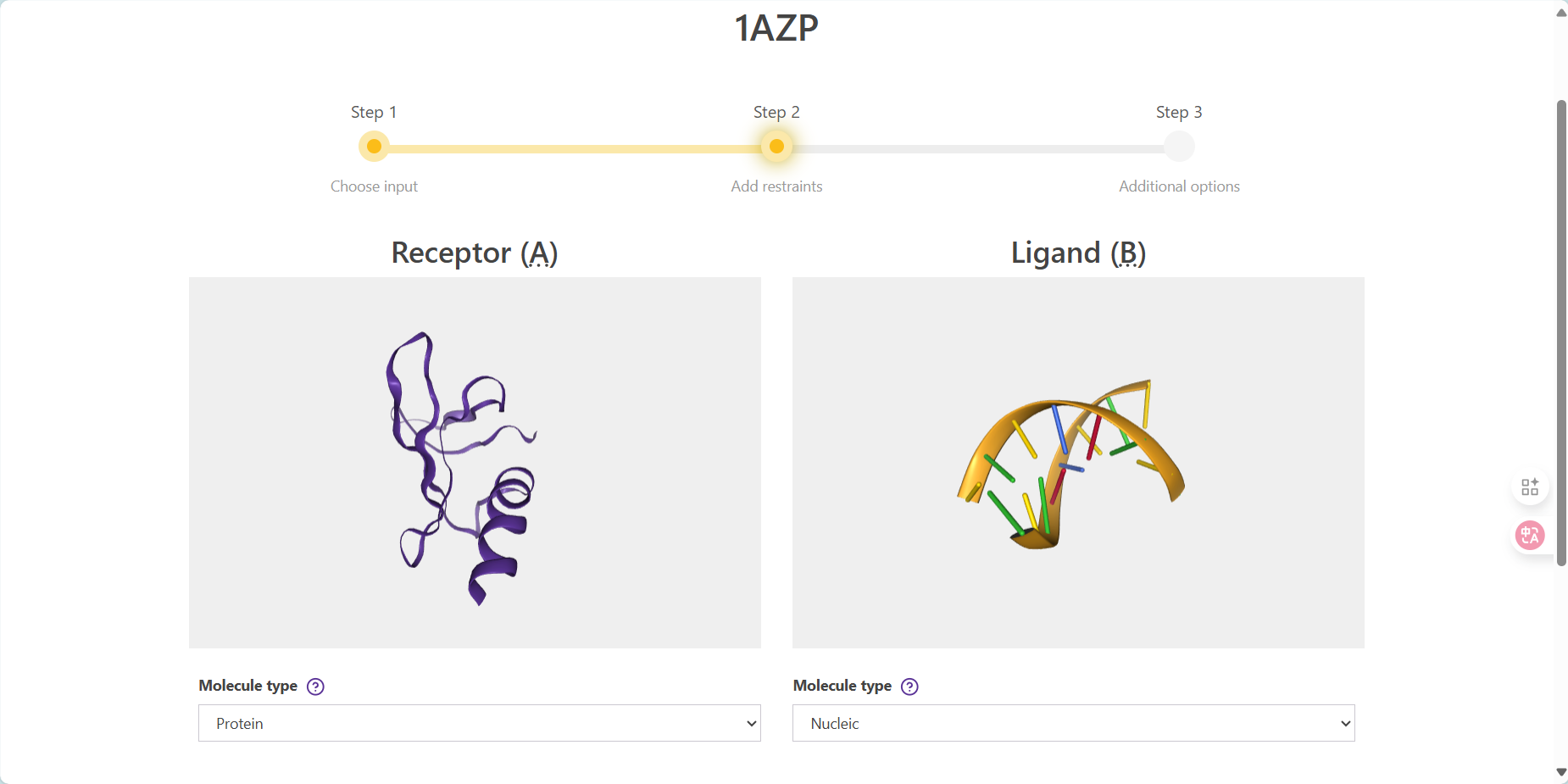
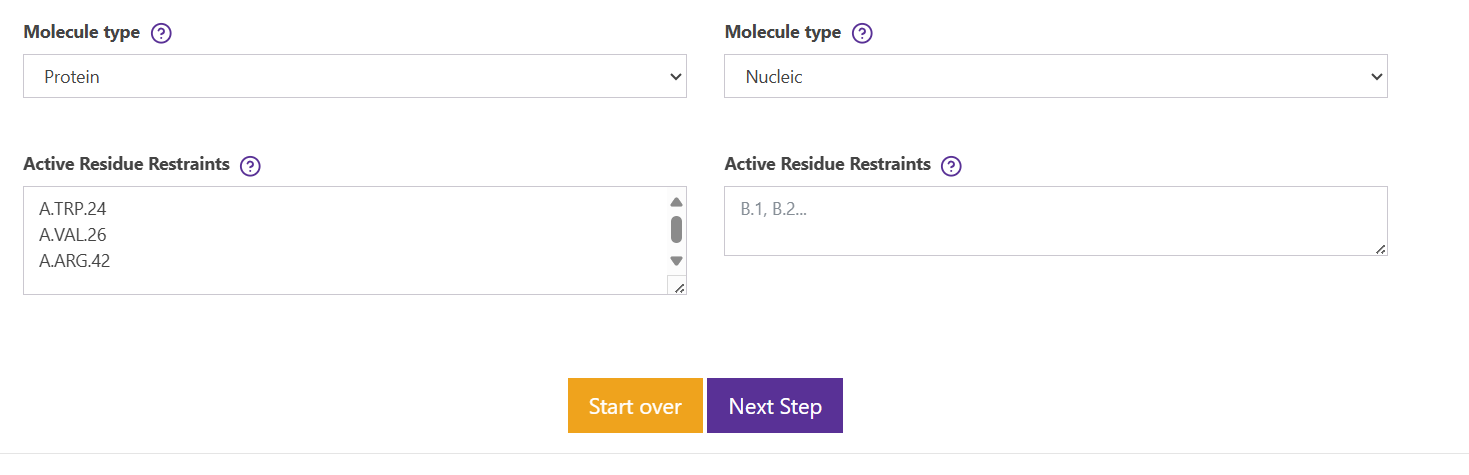
故去除约束 next step
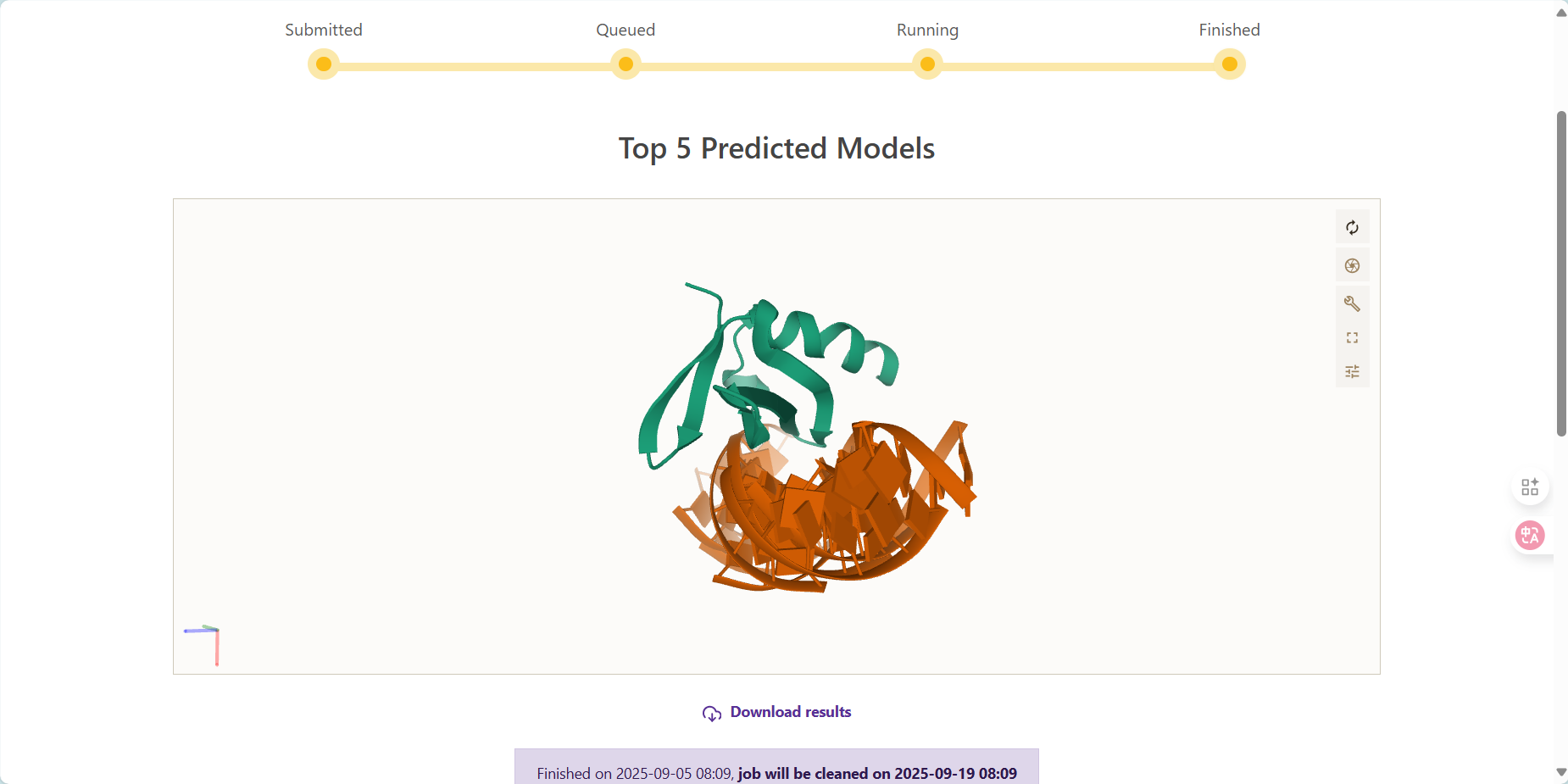
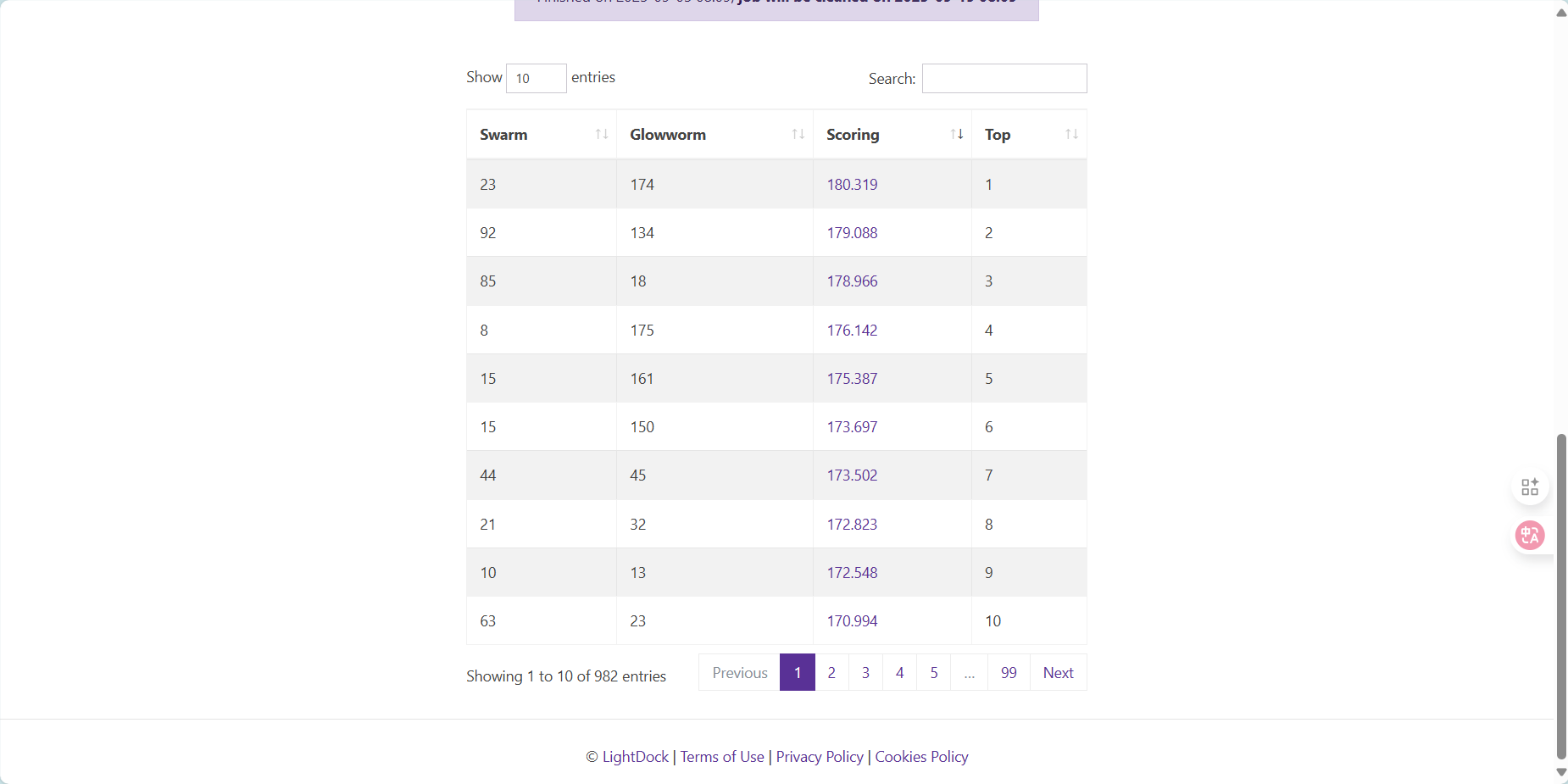
结果有正有负合理
2.常见警告⚠
Structure contains HETATM entries. Please remove them or rename to HETATM if possible.
蛋白pdb文件删除杂原子 HETATM;保留或去除水分子
Error reading provided PDB structure
结果:
DNA图像未出现
转换dome 可将DS visualizer .pdb 文件转换为 lightdock.server需要的文件
import sys
from pathlib import Path
def normalize_atom_name(raw_name: str) -> str:
name = raw_name.replace('"', '').strip()
# Rename phosphate oxygens to example style
if name == 'OP1':
name = 'O1P'
elif name == 'OP2':
name = 'O2P'
elif name == 'OP3':
# Example file does not use OP3; map to O3P if present
name = 'O3P'
return name
def format_atom_line(
serial: int,
name: str,
resname: str,
chain_id: str,
resseq: int,
inscode: str,
x: float,
y: float,
z: float,
) -> str:
# PDB fixed-width formatting approximating the example file layout
# Columns: Record(1-6), serial(7-11), atom name(13-16), resname(18-20), chain(22), resseq(23-26),
# coords x(31-38), y(39-46), z(47-54), occupancy(55-60), temp(61-66)
# Atom name must be right-justified in columns 13-16. Column 17 (altLoc) is a space.
name_field = name.rjust(4)[:4]
res_field = resname.rjust(3)[:3]
chain_field = (chain_id or 'B')[:1]
ins_field = (inscode or ' ')[:1]
return (
# Insert an explicit altLoc space between atom name and residue name
f"ATOM{serial:7d} {name_field} {res_field} {chain_field}{resseq:4d}{ins_field}"
f" {x:8.3f}{y:8.3f}{z:8.3f} 1.00 0.00"
)
def convert_pdb(in_path: Path, out_path: Path, target_chain: str = 'B') -> None:
lines_out = []
serial = 1
with in_path.open('r', encoding='utf-8', errors='ignore') as f:
for raw in f:
record = raw[:6].strip()
if record not in {"ATOM", "HETATM"}:
# Skip headers like REMARK, CRYST1, etc. Keep TER and END as we will add later.
continue
# Robustly parse using PDB column positions when available
# Fallback to whitespace splitting if line is too short
try:
name = raw[12:16]
resname = raw[17:20].strip() or 'UNK'
chain_id = target_chain or (raw[21:22].strip() or 'B')
resseq_str = raw[22:26].strip() or '0'
inscode = raw[26:27]
x = float(raw[30:38])
y = float(raw[38:46])
z = float(raw[46:54])
except Exception:
parts = raw.split()
if len(parts) < 8:
continue
# parts: ATOM serial name resname chain resseq x y z ...
name = parts[2]
resname = parts[3]
chain_id = target_chain or (parts[4] if len(parts) > 4 else 'B')
resseq_str = parts[5] if len(parts) > 5 else '0'
x, y, z = map(float, parts[6:9])
inscode = ' '
# Normalize fields
name = normalize_atom_name(name)
# Ensure sugar atom naming keeps apostrophes, e.g., O3', O5'
# Already handled by stripping quotes only
try:
resseq = int(resseq_str)
except Exception:
resseq = 0
# Build output line
out_line = format_atom_line(
serial=serial,
name=name,
resname=resname,
chain_id=target_chain or chain_id,
resseq=resseq,
inscode=inscode,
x=x,
y=y,
z=z,
)
lines_out.append(out_line)
serial += 1
# Append TER and END like the example
if lines_out:
# Try to infer last residue info for TER line formatting
last = lines_out[-1]
# Extract fields to place a TER similar to example (not strictly column-perfect)
ter_chain = (target_chain or 'B')[:1]
# Residue name and number from last line columns
resname = last[17:20]
resseq = last[22:26].strip()
lines_out.append(f"TER{''.ljust(8)} {resname} {ter_chain}{resseq:>4}")
lines_out.append("END")
out_path.write_text("\n".join(lines_out) + "\n", encoding='utf-8')
def main():
if len(sys.argv) < 3:
print(
"Usage: python convert_to_example_format.py <input_pdb> <output_pdb> [chain_id]",
)
sys.exit(1)
in_file = Path(sys.argv[1])
out_file = Path(sys.argv[2])
chain = sys.argv[3] if len(sys.argv) > 3 else 'B'
if not in_file.exists():
print(f"Input file not found: {in_file}")
sys.exit(1)
out_file.parent.mkdir(parents=True, exist_ok=True)
convert_pdb(in_file, out_file, target_chain=chain)
print(f"Converted {in_file.name} -> {out_file}")
if __name__ == '__main__':
main()
Flexible backbone (双√)
Flexible backbone 2 (单√2)
Flexible backbone 2 (单√1)
Linux
上传原始文件
Protonation 质子化
在生物化学和分子生物学中,质子化通常指的是分子或原子团接受一个质子(H+)的过程,这在蛋白质的结构和功能中尤为重要。
Protein
reduce -Trim 1AZP_A.pdb > 1AZP_A_noh.pdb
reduce -BUILD 1AZP_A_noh.pdb > 1AZP_A_h.pdb
DNA
reduce -Trim 1AZP_B.pdb > 1AZP_B_noh.pdb
reduce -BUILD 1AZP_B_noh.pdb > 1AZP_B_h.pdb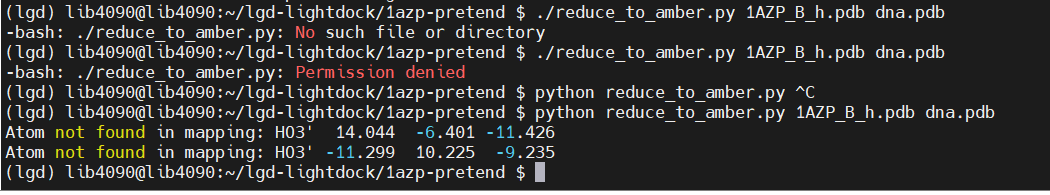
1.上传文件reduce_to_amber
python reduce_to_amber.py 1AZP_B_h.pdb dna.pdb3.Atom not found in mapping: HO3' 14.044 -6.401 -11.426
Atom not found in mapping: HO3' -11.299 10.225 -9.235
Setup
上传文件 restraints.list
lightdock3_setup.py protein.pdb dna.pdb -anm -rst restraints.list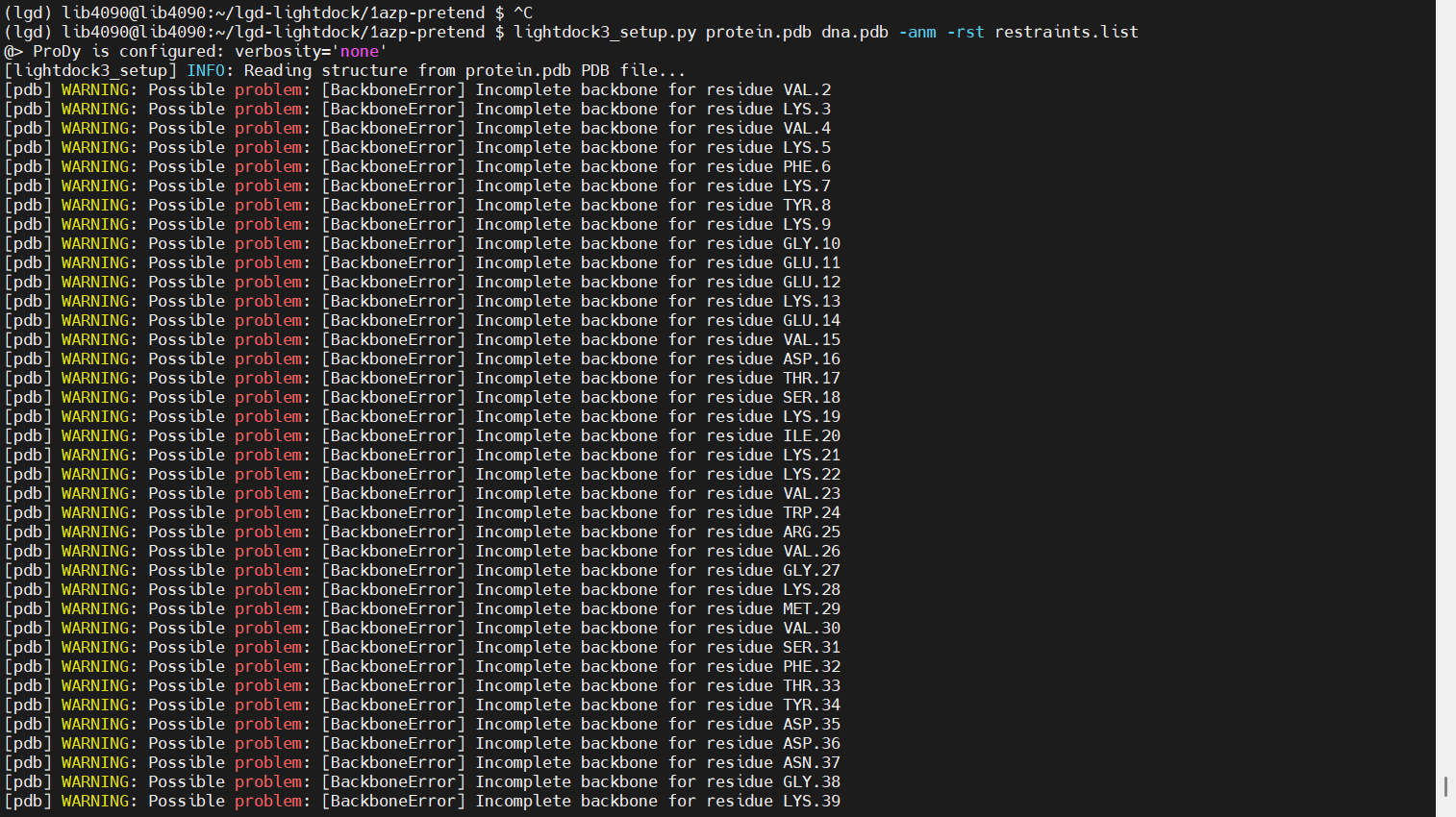
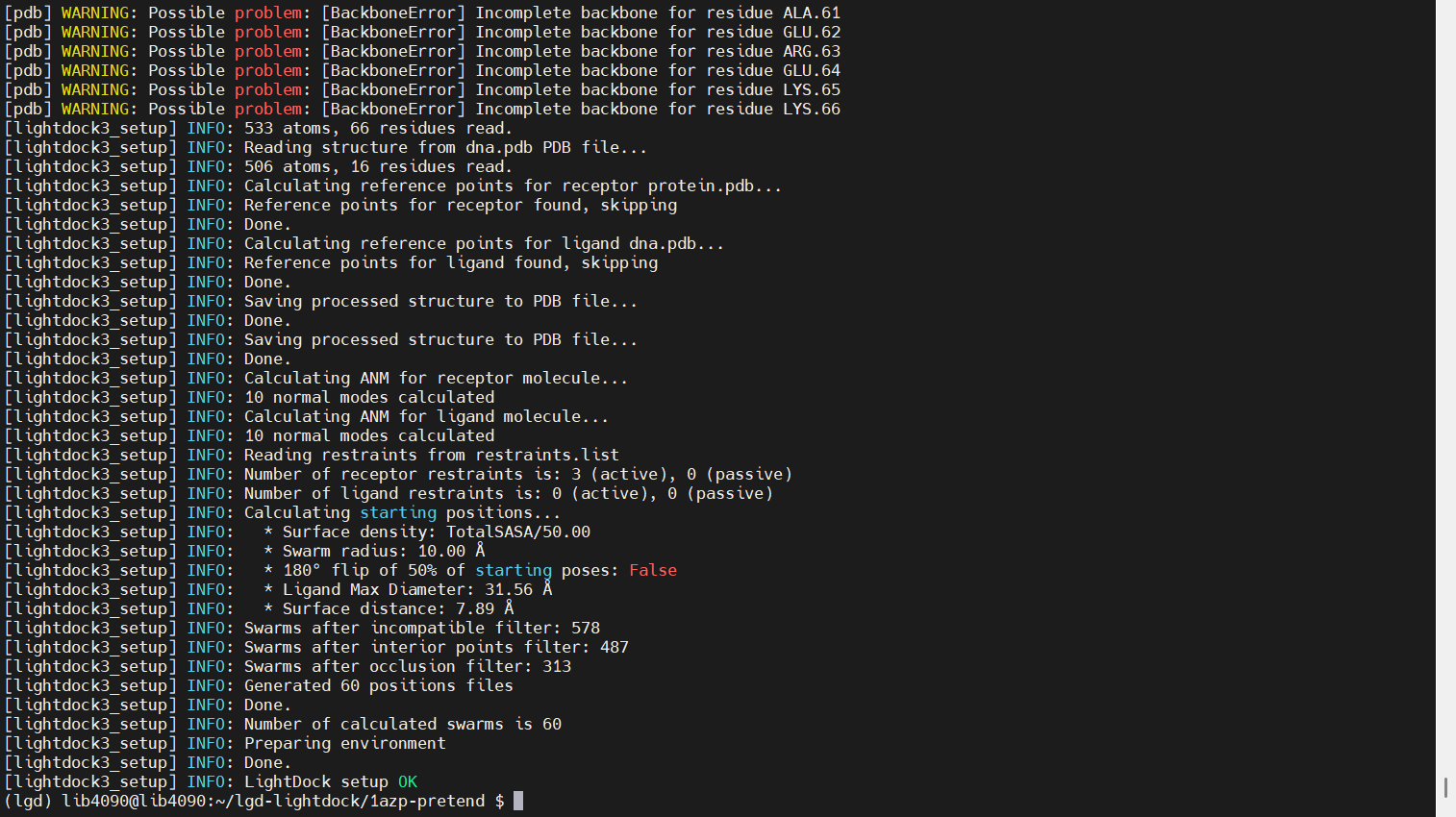
Simulation
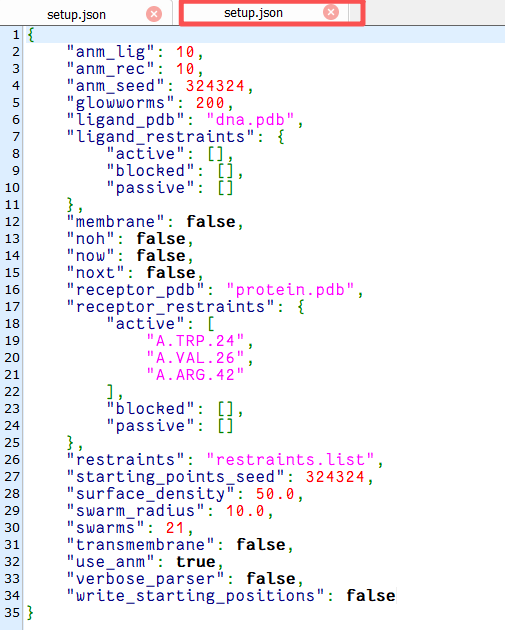
上传文件 setup.json
lightdock3.py setup.json 100 -s dna -c 8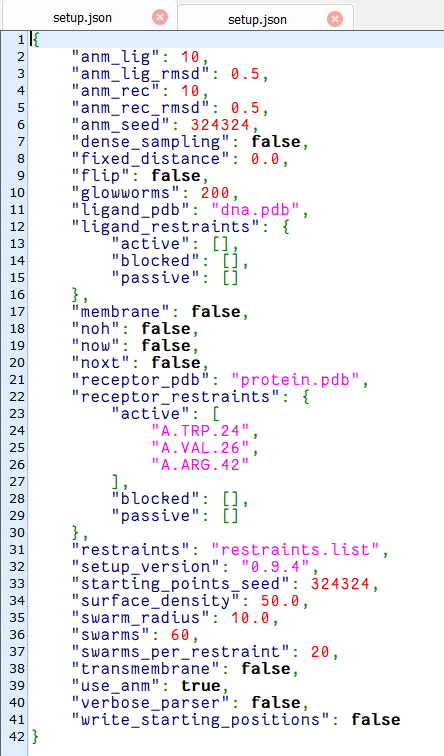
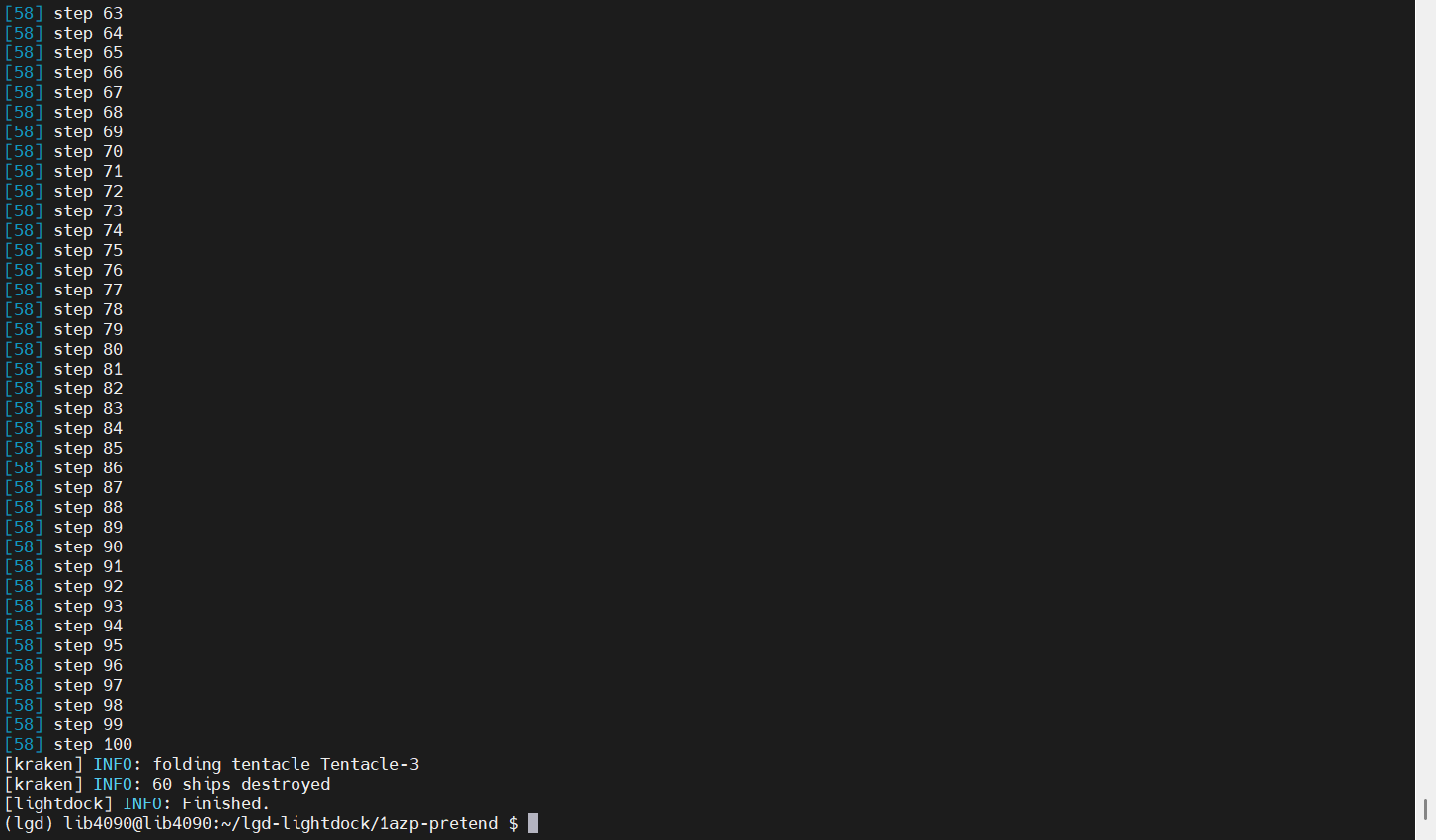
上传analyze.sh
./analyze.sh
















 2198
2198

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









