




上一期我们聊了归一化和高变基因筛选,为单细胞数据分析打下了坚实的基础。然而,你是否经历过这样的场景:满怀期待地绘制出UMAP,却发现不同样本的细胞仿佛说着不同语言,各自为政地分散成一座座孤岛?这正是批次效应在作怪!
本期我们将深入探讨批次效应的本质,梳理单细胞数据整合的主流方法,提供详细的工具选择指南,并结合R与Python双语代码示范,让你轻松驾驭数据整合这项关键技能。
1. 批次效应究竟是何方神圣?
1.1 什么是批次效应?
批次效应就像不同地方的方言,即使表达的内容相似,也会让人误以为是在讲不同的事情。在单细胞分析中,批次效应通常来源于实验流程差异、试剂批次、测序平台甚至操作者的不同。这些差异导致相同类型的细胞在表达数据中人为地产生偏移。
举个例子:假如两个实验室用不同的方法处理同一种细胞,Lab A细胞看上去稳定正常,Lab B细胞却呈现出高应激反应(如FOS/JUN的高表达),其实它们本质上是同一种细胞类型,但却被批次效应“强行”拆分成了两个群体。
1.2 不做数据整合,危害到底有多大?
批次效应对后续分析会产生非常严重的干扰:
| 后续分析步骤 | 受到的影响 |
|---|---|
| 降维可视化 | UMAP图中,细胞会按照实验批次而不是细胞类型聚集 |
| 聚类分析 | 同种细胞可能被强行拆散,按照实验批次重新聚类 |
| 差异基因分析 | 批次相关的技术噪声基因被错误标记为生物差异基因 |
| 轨迹推断与细胞通讯分析 | 伪造的差异导致轨迹分析不连续,细胞通讯分析也出现虚假结果 |
因此,整合批次效应势在必行。不整合,就像用不同的语言强行沟通,结果必然是一片混乱。
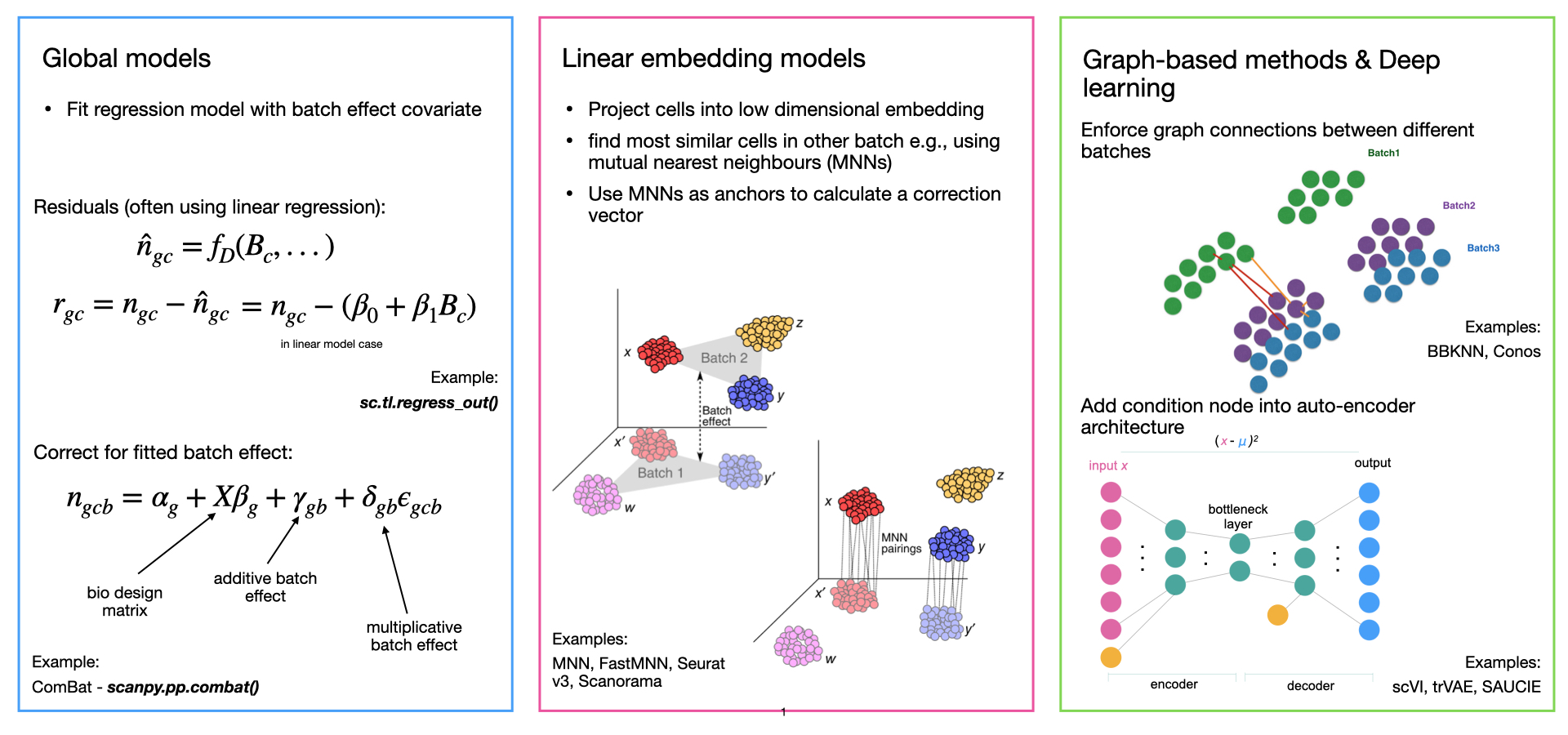
2. 数据整合的方法分类与选择指南
单细胞数据整合方法主要可以归纳为以下四大家族,每种方法都有独特的适用场景与局限。
| 方法类别 | 通俗理解 | 常用工具(R) | 常用工具(Python) | 优势与不足 | 推荐场景 |
|---|---|---|---|---|---|
| 全局模型 | 类似照片整体调亮,简单直接 | ComBat | scanpy.pp.combat | 快速易用,适合轻度批次差异,但难以处理复杂差异 | 小数据、技术重复数据 |
| 线性嵌入模型 | 通过数学变换与微调局部差异,好比精细调色 | Seurat(CCA)、Harmony、FastMNN | Scanorama、Harmony-py | 易用性强,适合中等规模数据,CPU运行即可 | 常规单细胞实验 |
| 图基模型 | 强制构建不同批次间的友谊桥梁 | - | BBKNN | 快速构建邻居图,直观简单,但整合效果较粗糙 | 多批次快速分析 |
| 深度学习模型 | 就像精通多国语言的同声传译,强大而灵活 | scVI/scANVI | scVI/scANVI/scGen | 处理能力强,能整合非线性差异,但需GPU支持与复杂调参 | 大规模、跨平台、多模态整合 |
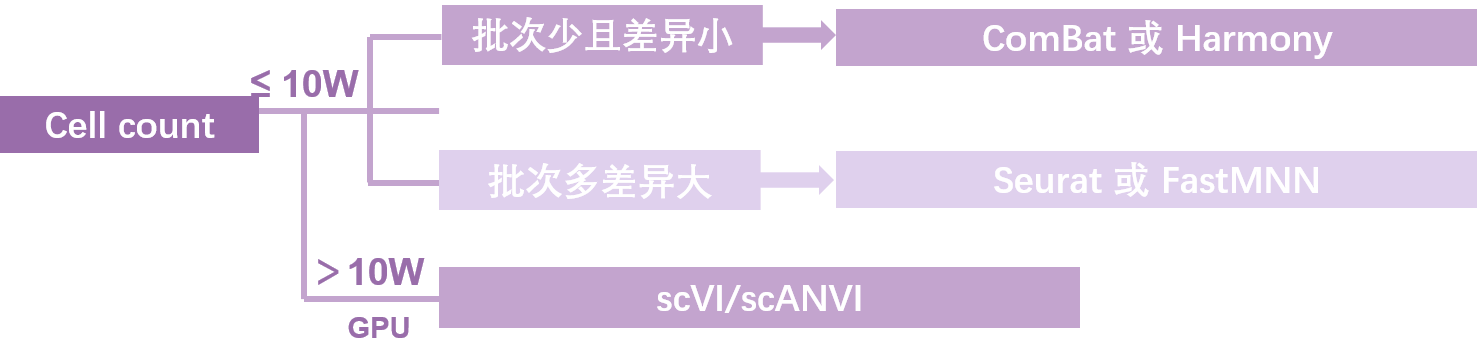
如何选择适合你的方法?参考以下流程图:
3. 整合效果如何评估?
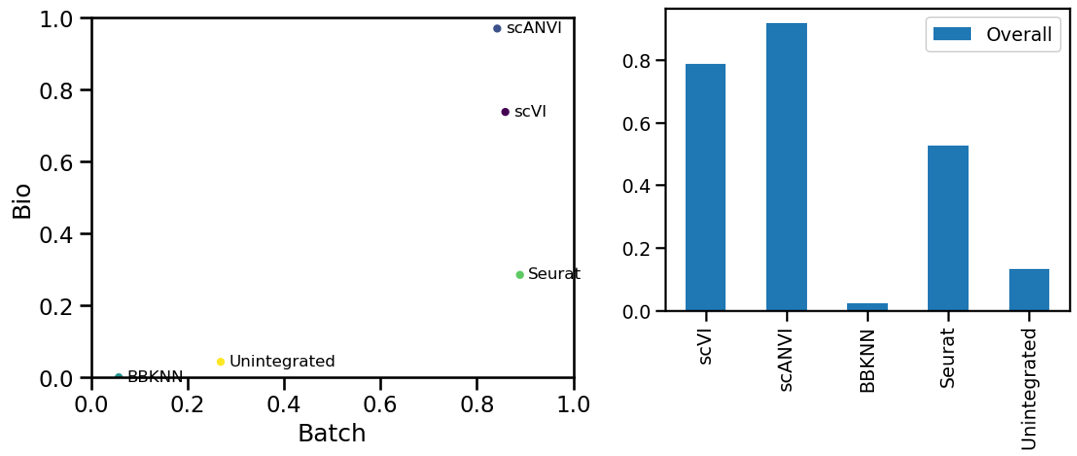
方法选好了,但效果如何客观评估呢?我们有几个常用的评估指标:
| 指标 | 含义 | 理想表现 |
|---|---|---|
| kBET/iLISI | 测量细胞在低维空间的批次混合程度 | 越高越好 |
| cLISI/Silhouette | 测量生物学信号是否被保留 | 越高越好 |
| Graph connectivity | 衡量图结构连接的稳定性 | 中高 |
| ASW_batch | 综合考量批次与生物信号 | 折中 |
同时推荐可视化检查:
-
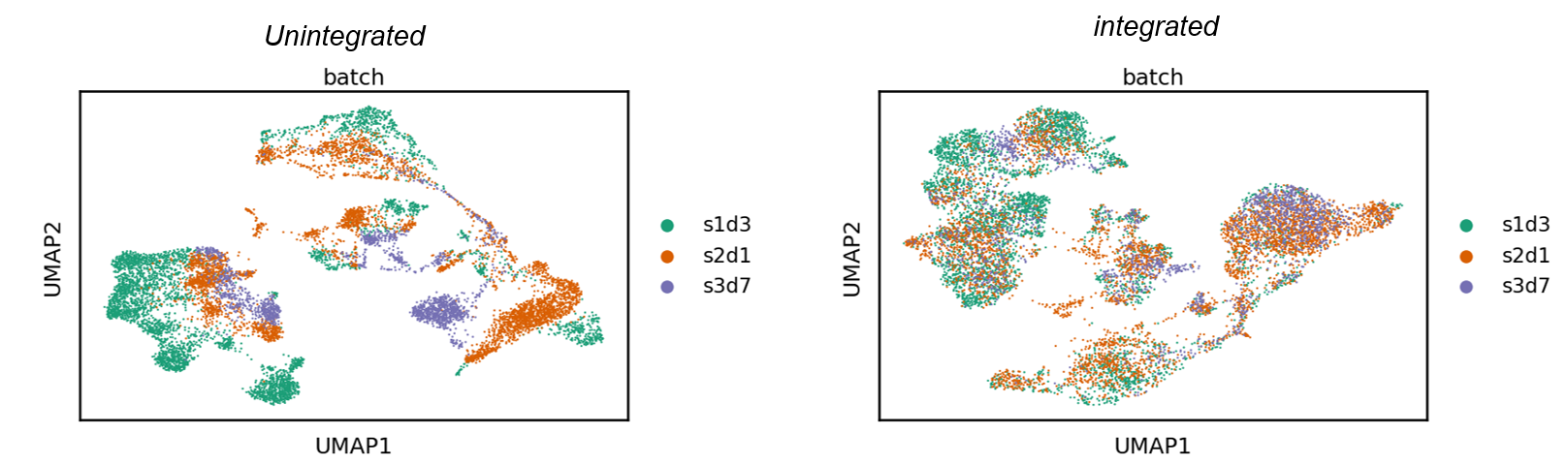
批次vs细胞类型的双UMAP图:好坏一目了然。
-
评估指标条形图:不同方法横向对比,高效展示效果。
4. 实操演练:代码模板(R & Python)
R语言(Seurat + Harmony)
library(Seurat)
# SCTransform归一化
obj.list <- SplitObject(seu, split.by = "batch") %>%
lapply(SCTransform, vars.to.regress = "percent.mt")
anchors <- FindIntegrationAnchors(obj.list, normalization.method = "SCT")
seu.int <- IntegrateData(anchors)
# Harmony整合(进一步优化)
seu.int <- RunPCA(seu.int) %>%
RunHarmony("batch") %>%
RunUMAP(reduction = "harmony", dims = 1:30)
🌱R中使用scVI
library(Seurat)
library(reticulate)
# ⚠️ 如果你还未指定conda环境,请确保已创建好一个名为scanpy的conda环境
use_condaenv("scanpy", required = TRUE)
#【0】(可选)PCA初步降维检查(为整合前检查数据结构)
seurat_obj <- RunPCA(seurat_obj, features = VariableFeatures(seurat_obj), reduction.name = "pca")
ElbowPlot(seurat_obj)
DimPlot(seurat_obj, reduction = "pca", group.by = "orig.ident")
#【1】整合前UMAP(未整合的数据情况对比)
seurat_obj <- RunUMAP(seurat_obj, reduction = "pca", dims = 1:30, reduction.name = "umap_unintegrated")
DimPlot(seurat_obj, reduction = "umap_unintegrated", group.by = "orig.ident")
#【2】⭐ 调用 scVI 进行数据整合(核心代码)
seurat_scvi <- IntegrateLayers(
object = seurat_obj, # 此处替换为你自己的Seurat对象
method = scVIIntegration,
new.reduction = "integrated.scvi",
conda_env = "scanpy",
verbose = FALSE
)
#【3】UMAP可视化整合后的数据
seurat_scvi <- RunUMAP(seurat_scvi, reduction = "integrated.scvi", dims = 1:20, reduction.name = "umap.scvi")
#【4】整合后的聚类(推荐)
seurat_scvi <- FindNeighbors(seurat_scvi, reduction = "integrated.scvi", dims = 1:20)
seurat_scvi <- FindClusters(seurat_scvi, resolution = 0.3, cluster.name = "scvi_clusters")
#【5】结果展示:整合后的UMAP
DimPlot(seurat_scvi, reduction = "umap.scvi", label = TRUE, split.by = "orig.ident") +
ggtitle("scVI Integrated UMAP")
⚙️ 使用说明:
-
将上述代码中的
seurat_obj替换为你自己的Seurat对象名称。 -
确保已通过
reticulate包连接到安装有scanpy和scVI的conda环境。 -
IntegrateLayers是Seurat v5支持的调用scVI的标准方法,请确认Seurat版本≥5。
🎯 最佳实践建议:
-
整合前务必确认所有样本的数据已统一经过归一化(Normalization) 和 高变基因(Variable Features)筛选,确保整合的效果最佳。
-
批次效应强烈时,适当增加
dims数目(如dims=1:30)可能提升效果。 -
UMAP的维度数(如
dims=1:20)建议根据ElbowPlot结果选择。
🛠️ 常见坑排查:
-
若出现reticulate环境报错,请检查
use_condaenv("scanpy")中conda环境名称是否与你电脑上的一致。 -
scVI运行缓慢时,建议检查你的conda环境中PyTorch是否已安装GPU版本。
Python语言(Scanpy + scVI)
#依赖库导入与设置环境import scanpy as sc
import scvi
import bbknn
import anndata as ad
# 如果使用R和Python混合环境,需要提前设置环境路径(可选)
import os
os.environ["R_HOME"] = r"<your-R-path>"
os.environ["R_USER"] = r"<your-rpy2-path>"
#合并数据并定义批次(batch)
# 假设 adata1 和 adata2 是你的数据对象
adata1.obs["batch"] = "batch1"
adata2.obs["batch"] = "batch2"
# 合并数据
adata_combined = ad.concat([adata1, adata2])
#整合方法一:使用Combat(适用于简单线性整合场景)
sc.pp.combat(adata_combined, key='batch')
#整合方法二:使用scVI深度整合(推荐,更强大且适用于大数据)
# 配置scVI整合
scvi.model.SCVI.setup_anndata(adata_combined, batch_key="batch")
# 构建并训练scVI模型
model = scvi.model.SCVI(adata_combined)
model.train()
# 提取整合后的低维空间
adata_combined.obsm["X_scVI"] = model.get_latent_representation()
# UMAP可视化
sc.pp.neighbors(adata_combined, use_rep="X_scVI")
sc.tl.umap(adata_combined)
sc.pl.umap(adata_combined, color="batch")
#整合方法三:BBKNN图整合(适用于大规模快速初筛)
sc.pp.pca(adata_combined)
bbknn.bbknn(adata_combined, batch_key="batch")
sc.tl.umap(adata_combined)
sc.pl.umap(adata_combined, color="batch")
#UMAP批次密度图(评估整合效果)
sc.tl.embedding_density(adata_combined, groupby="batch")
sc.pl.embedding_density(adata_combined, groupby="batch")
#整合效果的细胞比例对比(可视化不同batch中各cluster比例)
# 首先确保已完成聚类
# sc.tl.leiden(adata_combined, resolution=0.5, key_added='cluster')
import pandas as pd
import matplotlib.pyplot as plt
# 计算各batch下每个cluster的细胞数量
cell_counts = adata_combined.obs.groupby(['batch', 'cluster']).size().reset_index(name='cell_count')
# 计算比例
cell_counts['total_cells_in_batch'] = cell_counts.groupby('batch')['cell_count'].transform('sum')
cell_counts['proportion'] = cell_counts['cell_count'] / cell_counts['total_cells_in_batch']
# 数据重塑用于绘图
proportions_pivot = cell_counts.pivot(index='batch', columns='cluster', values='proportion')
# 绘制堆叠比例图
proportions_pivot.plot(kind='bar', stacked=True, figsize=(12, 8))
plt.title('Proportion of Each Cluster by Batch')
plt.ylabel('Proportion')
plt.xlabel('Batch')
plt.legend(title='Cluster', bbox_to_anchor=(1.05, 1), loc='upper left')
plt.tight_layout()
plt.show()
📌 使用说明与注意事项
-
将模板中
adata1、adata2替换成自己的数据对象。 -
如果你的数据批次更多,只需按模板扩展
ad.concat([...])即可。 -
可根据需求灵活选择整合方法:
-
ComBat:小规模线性批次整合。
-
scVI:深度学习强整合,推荐。
-
BBKNN:快速粗略整合,适合大规模数据的初步分析。
-
5. 常见问题与避坑指南(FAQ)
| 症状 | 可能原因 | 急救措施 |
|---|---|---|
| UMAP 仍分批 | 变量基因集不一致 | 统一 genes_to_integrate 或 SCT pipeline |
| scVI loss 不收敛 | 学习率过低 / 批次超多 | lr↑、增 n_latent、梯度裁剪 |
| BBKNN 后聚类“碎片化” | 稀有细胞被过分强连边 | 调整 perturbations=True 或后做 Leiden resolution↓ |
6. 延伸阅读 & 资源汇总
-
Benchmark:Luecken et al. 2021 Nature Methods——14 个指标评测 16 种方法。sc-best-practices.org
-
评价工具:
scIB、batchbenchpipeline,支持一键跑 kBET/iLISI/ASW。sc-best-practices.org -
多模态整合:WNN (Seurat v5)、GLUE、totalVI 教程见 sc-best-practices paired integration 章节。sc-best-practices.org
7. 小结 & 下期预告
-
批次效应的本质是“技术方言”;整合就是训练“同声传译”。
-
四大方法家族各擅胜场,没有“一招鲜”,结合样本规模与硬件选择最重要。
-
评估时要兼顾“批次混合”与“生物保真”,kBET / iLISI + Silhouette 是黄金搭档。
-
R 与 Python 生态均有成熟代码模板,牢记 预处理一致、变量基因同步、指标量化 三大关键。
下一期我们将深入降维算法背后的数学直觉,比较 PCA / UMAP / t-SNE,并实战 Leiden / Louvain 聚类调参,带你把整合后的坐标系“切片”成清晰的细胞类型地图。敬请期待!



















 9591
9591

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











