




单细胞测序数据分析领域有一句经典的话:“语言和生态的选择,决定了你的上限。”当我们深入到数据质控的细节时,这句话尤为贴切。无论你是Seurat(R)忠粉,还是Scanpy(Python)拥趸,都会遇到生态壁垒和数据流通问题。更何况,数据质量本身的判断,也是一门细致活。
这一期,我们将系统梳理R/Python两大阵营的数据结构、互操作技巧,以及单细胞数据质控的全部流程。每个环节都力求“知其然,更知其所以然”
1. Seurat与Scanpy数据结构
1.1 Seurat对象结构与生态
Seurat对象,可以理解为一个精致的“多层蛋糕”,每一层都装载着不同维度的信息。最常见的数据结构包括:
- assays:存储原始计数(counts)、归一化数据(data)、标准化数据(scale.data)
- meta.data:每个细胞的“档案袋”,记录细胞类型、批次、线粒体比例等
- reductions:降维结果(如PCA、UMAP坐标)
- graphs:图结构,用于聚类等操作
这一切都被包装在一个Seurat对象里。比如你用Read10X()加载10X的原始mtx文件后,得到的就是这样一个对象,可以方便地用object@meta.data查看细胞属性,或用object@assays$RNA@counts读取表达矩阵。
适用场景
Seurat极其适合数据预处理、降维、聚类和可视化,尤其是在R环境下与Bioconductor深度兼容。其图形可视化能力强,生态繁荣,适合交互式探索。
安装与基础操作示例
# 安装
install.packages("Seurat")
library(Seurat)
# 数据读取
seurat_obj <- Read10X(data.dir = "filtered_feature_bc_matrix/")
# 查看meta data
head(seurat_obj@meta.data)
1.2 Scanpy/AnnData对象结构与生态
- AnnData 是 scverse (Python) 生态的核心类。它把转录本计数矩阵放在 .X,维度为 obs × var:行是细胞条形码,列是基因。围绕这块主矩阵,它再用 6 对“抽屉”组织附加信息:
| 抽屉 | 尺寸 | 典型内容 |
|---|---|---|
obs / var | n_obs × p / n_var × q | 细胞/基因注释 (batch、gene_id…) |
obsm / varm | n_obs × k / n_var × k | UMAP 坐标、周期得分、载荷 |
obsp / varp | n_obs × n_obs | KNN 或共表达图 |
layers | 与 .X 同形 | 原始 counts、归一化、imputed |
uns | 任意 | 配色、参数字典等非结构化信息 |
AnnData 对象通常由 scanpy.read_10x_mtx()、sc.read_h5ad() 或直接用稀疏矩阵初始化;写盘时会序列化为 H5AD——一种 HDF5 层级存储,能在磁盘上保持 AnnData 的“抽屉结构”
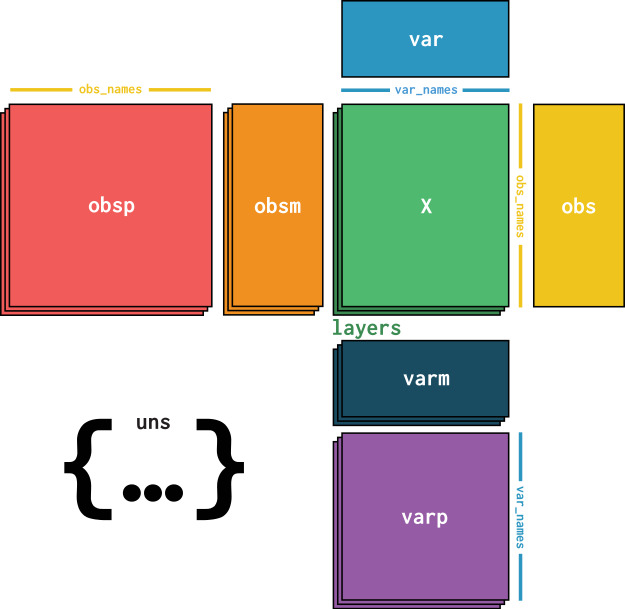
Fig. 4.1 AnnData overview. Image obtained from [Virshup et al., 2021].
举例来说,当你用sc.read_10x_mtx()加载mtx数据时,AnnData对象的.X属性就是主表达矩阵,.obs就是细胞的注释信息。
适用场景
Scanpy更倾向于大数据集的高效批量处理,适合脚本化自动化流程、批量分析,也有越来越多的机器学习和多模态集成新工具(如scvi-tools、muon)。
# 安装
pip install scanpy
import scanpy as sc
# 数据读取
adata = sc.read_10x_mtx('filtered_feature_bc_matrix/', var_names='gene_symbols')
# 查看meta data
adata.obs.head()
1.3 Seurat与Scanpy的异同点
| 属性 | Seurat(R) | Scanpy/AnnData(Python) |
|---|---|---|
| 数据结构 | S4对象、slot式 | AnnData类、dict/np结构 |
| 元数据 | meta.data | obs |
| 降维结果 | reductions | obsm |
| 生态扩展 | 强,Bioconductor丰富 | 快速发展,ML集成多 |
| 可视化 | 强,交互丰富 | 脚本化为主,图表可自定制 |
| 数据体量 | 支持中等~大数据 | 更适合超大规模、批处理 |
打个比方:Seurat像是“Excel + 强大制图工具”,Scanpy更像“Pandas + Numpy + 自动化脚本”,各自有各自的“舒适圈”。
2. R与Python生态系统切换与互操作性
2.1 单细胞分析三大生态系统
-
R:Bioconductor(包罗万象)、Seurat(专注单细胞)
-
Python:scverse(scanpy为核心,延展到多模态、机器学习)
什么时候需要切换?
- 某些高分辨率批量处理,Python更高效
- 需要用到特定算法(如scVI、Harmony等),可能只在一边实现
- 想结合R的可视化和Python的批量脚本处理
2.2 命名法差异
- R中习惯用驼峰(如meta.data),Python多用下划线(如obs_names)
- 代码风格会影响阅读体验,需注意转换
2.3 基于磁盘的互操作性
原理:把对象保存成标准格式文件(如.h5ad、.loom、.rds),然后另一语言读取
优缺点:存储空间大、步骤稳定,适合流水线,但交互性较差,像是寄一张明信片:写-读稳定,但每换一次车都得再投邮筒
常见格式及互通方法
| 格式 | 来源 | 生成方式 | 适用场景 |
|---|---|---|---|
| H5AD | anndata | adata.write_h5ad("file.h5ad") 或 Convert(..., dest="h5ad") | Python ↔ R 最主流;支持稀疏块读写、压缩 |
| LOOM | loompy 规范 | loompy.create(...) / connect() (R: loomR) | 与生态无关,RNA-velocity 等工具默认输出 |
| RDS | base R | saveRDS(seu,"obj.rds") | 只在 R 内部快速存档;跨语言可读性差,公开数据不推荐 |
💡Tip 写 H5AD 时,务必告知 X 是转置过的 counts,否则读取后会出现“行列颠倒”惊悚片
2.4 基于内存的互操作性
reticulate (R 调 Python)、rpy2 (Python 调 R)。实时、交互,但版本依赖重、易占双倍内存。像是共享白板,实时但对版本与依赖挑剔
内存互通模板
# Python 调 R :rpy2
import rpy2.robjects as ro
from rpy2.robjects import pandas2ri
pandas2ri.activate()
ro.r('library(Seurat)')
seu = ro.r('CreateSeuratObject')(mat) # 直接调用 R 函数
# R 调 Python :reticulate
library(reticulate)
sc <- import("scanpy")
adata <- sc$read_h5ad("sample.h5ad")
3. 单细胞数据质量控制(QC):理论基础与核心流程
3.1 质控的科学依据与核心目标
单细胞转录组测序(scRNA-seq)技术在揭示细胞异质性方面具有独特优势,但其数据易受多种技术与生物学因素影响,因此系统的质量控制(QC)环节是保障后续分析可靠性的前提。
QC 的核心目标是剔除技术噪声、非真实生物信号,从而确保每个被分析的细胞都具有生物学意义和高数据质量。具体需要应对的问题包括:
-
高水平的技术噪声与掉点(dropout):由于测序深度有限和捕获效率低,部分细胞表达谱极度稀疏,许多真实表达基因可能测不到。
-
不均一的细胞完整性:部分细胞可能因膜破裂、RNA 降解等原因表现出极低的分子计数或高比例的线粒体基因表达。
-
批次效应与污染信号:不同样本、文库或测序批次间的技术差异,以及实验体系中的环境 RNA 污染,会进一步影响数据的真实性和可比性。
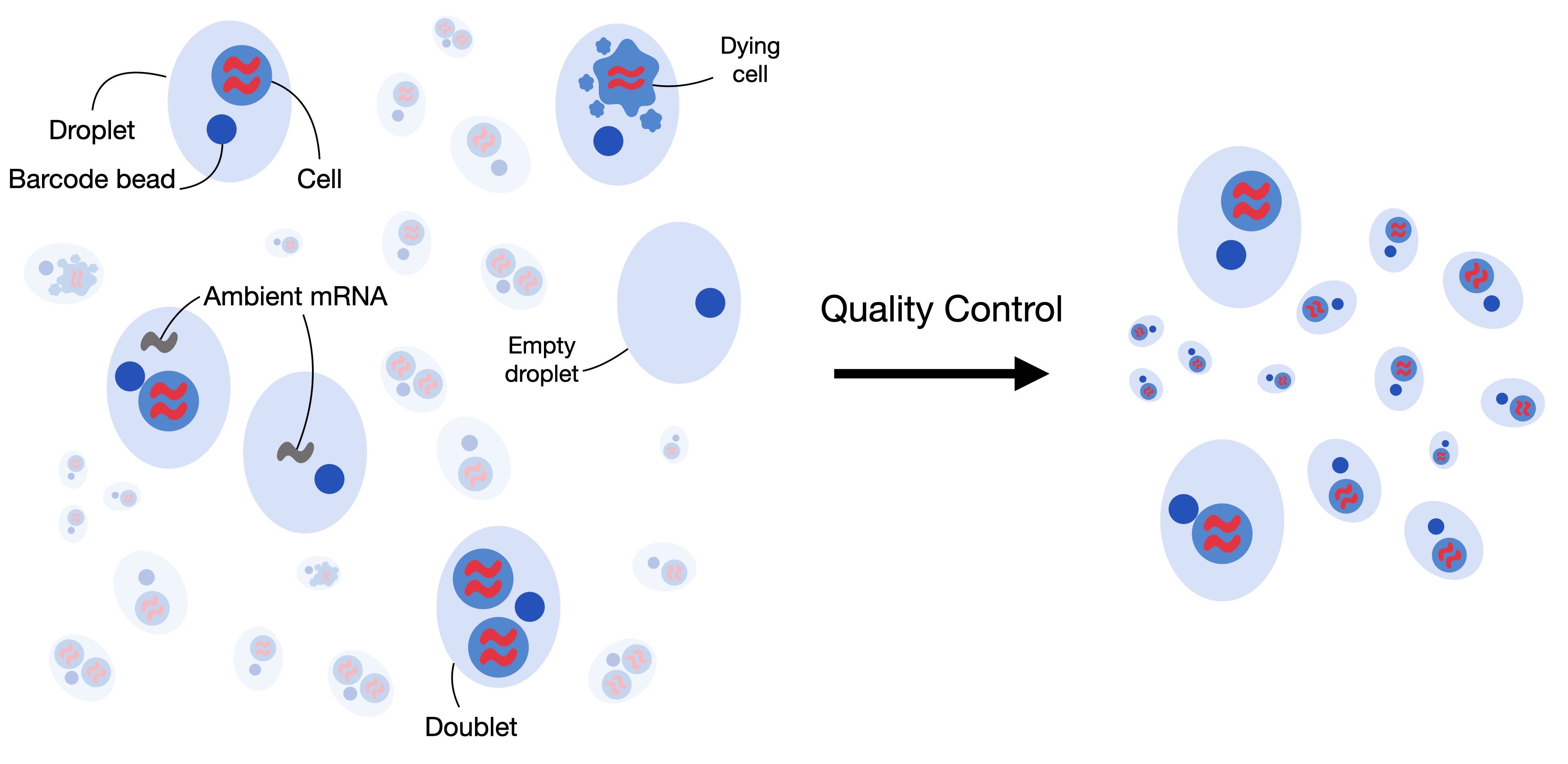
Fig.4.2 scRNA-seq datasets can contain low-quality cells, cell-free RNA and doublets
因此,科学的 QC 流程需针对以上问题设置多重过滤和校正策略,剔除质量不合格的细胞,为后续分析提供高可信度的数据集。
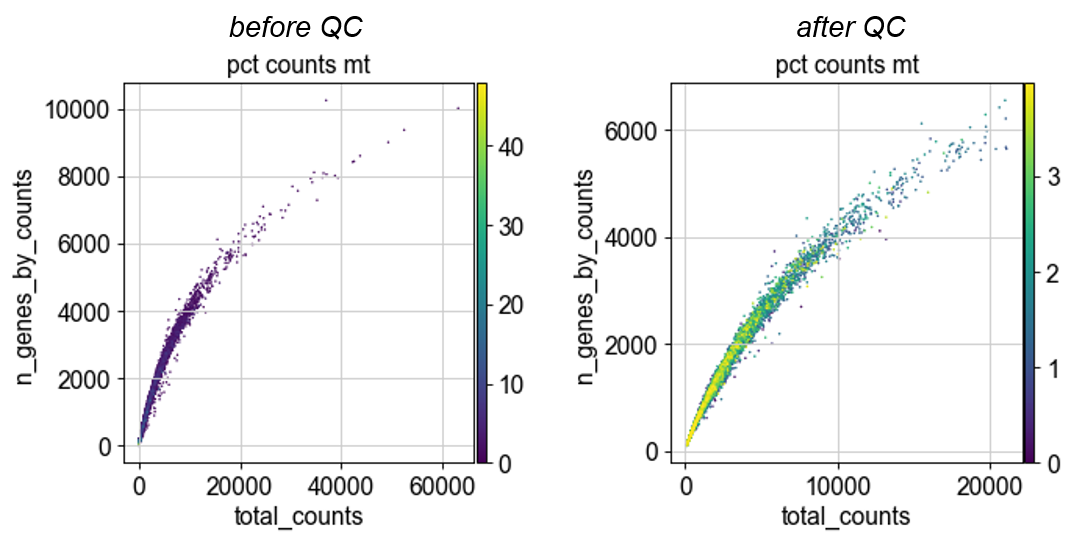
3.2 QC 步骤、关键指标与常用阈值
核心 QC 步骤包括:
| 步骤 | 目标 | 关键指标(英文) | 典型阈值 | 解释与意义 |
|---|---|---|---|---|
| 1. 评估细胞活性 | 剔除低活性或损伤细胞 | n_genes_by_counts (nFeature) | > 200–500 | 反映细胞的转录活性与完整性;过低提示细胞破损或空滴 |
| 2. 去除高分子计数异常细胞 | 排除潜在双细胞或异常大细胞 | total_counts (nCount) | < 50,000–100,000 | 过高可能是两个细胞共用 barcode,或异常高表达 |
| 3. 线粒体基因比例过滤 | 剔除死亡/应激细胞 | pct_counts_mt (percent.mt) | < 10–20% | 线粒体基因表达高通常提示细胞死亡或应激状态 |
| 4. 基因过滤 | 保证每个基因在一定数量细胞中被检测 | min_cells | > 3 | 极低检测基因多为背景或技术噪声 |
这些过滤步骤的阈值应结合物种、组织类型和实验设计灵活调整。建议在实施前先用小提琴图、散点图等方式整体可视化各指标分布,以指导合理的参数选择。
3.3 RNA 污染(Ambient RNA)校正
在 droplet-based scRNA-seq 技术中,环境中的游离 RNA(ambient RNA)会被液滴系统捕获并计入到细胞表达中,导致假阳性的低水平表达,尤其在低 UMI 细胞中更为显著。未经处理的污染信号会干扰细胞类型鉴定和后续生物学解释。
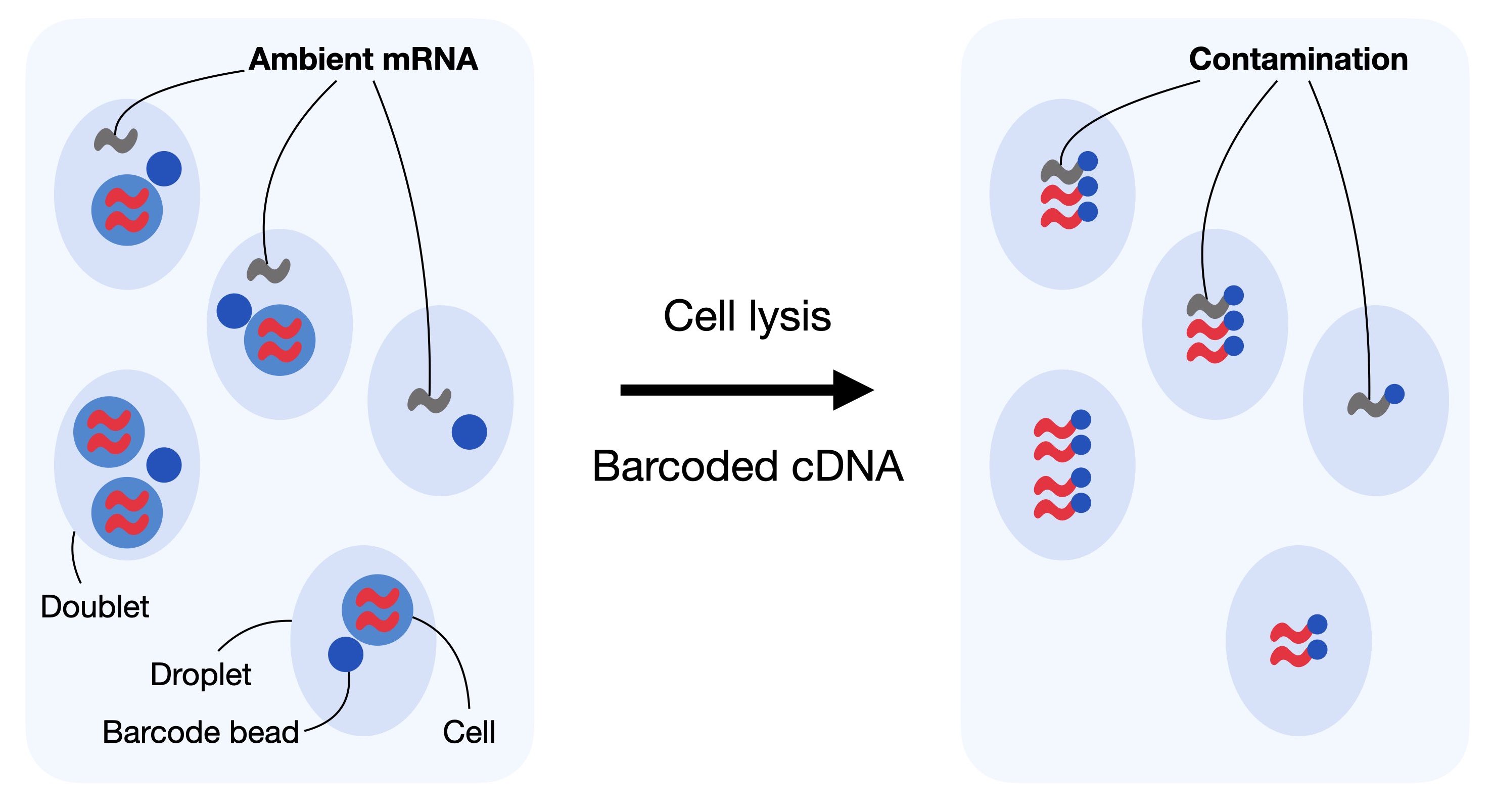
Fig.4.3 droplets can incorporate ambient RNA or doublets
常用校正方法包括 SoupX 和 DecontX:
-
SoupX:基于背景表达谱估算污染比例,并从表达矩阵中扣除推测的污染计数。适用于10X Genomics等 droplet 平台,操作简便,且与 Seurat 高度兼容。
-
DecontX:采用贝叶斯层级模型,根据细胞分群结果估计污染比例,具有更强的灵活性,可直接在 R/Python 端对稀疏计数矩阵运行。
污染校正后,建议重新评估整体表达分布,并结合污染比例分布对细胞进行再次筛选,确保数据真实性。
3.4 双细胞(Doublet)检测
微流控捕获的随机性会导致少量条形码包含两个或多个细胞,即“双细胞”现象。双细胞表达矩阵的混合特征会伪造稀有亚群、影响聚类结构甚至干扰差异分析。因此,检测并剔除双细胞是标准的 QC 流程之一。
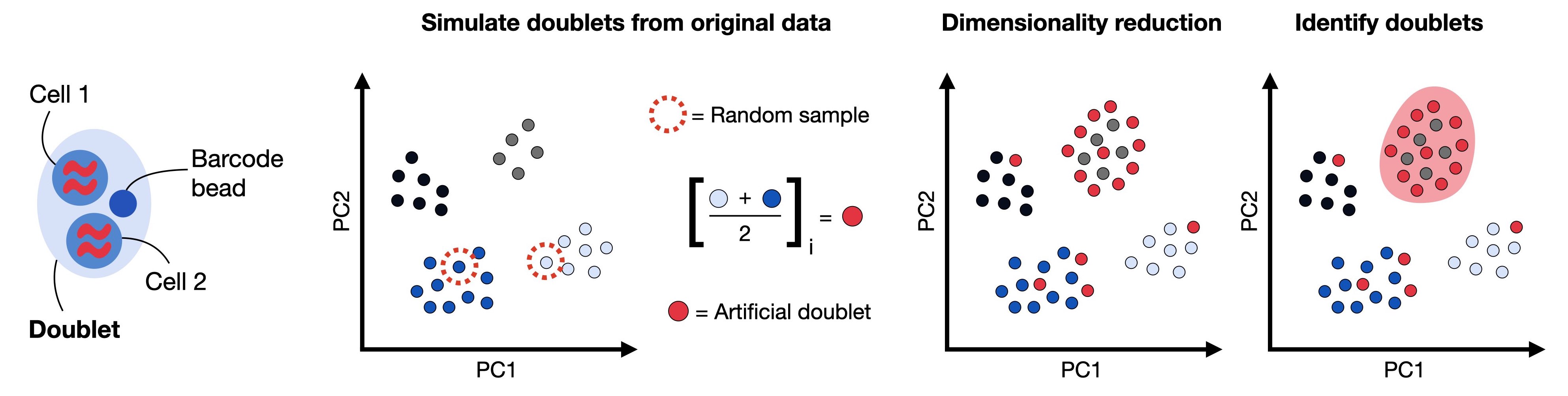
Fig. 4.4 Doublets are droplets that contain more than one cell.
主流检测方法基于模拟混合表达分布、KNN 最近邻评分或机器学习分类,常用工具包括:
| 工具 | 生态 | 原理 | 应用要点 |
|---|---|---|---|
| DoubletFinder | R | 模拟混合分布 | Seurat 对象直接支持 |
| scDblFinder | R | 自动聚类-混合 | 兼容 Bioconductor 框架 |
| Scrublet | Python | PCA + KNN评分 | 适用于大规模高通量 |
检测后,应根据双细胞概率分布或设定阈值剔除高概率条形码,并在降维结果上验证其空间分布,确保不会错误移除真实稀有亚群。
3.5 QC 步骤与合理性评判
每一步 QC 的阈值设置都应基于实验背景、数据分布和生物学预期动态调整。
过严的阈值会降低数据多样性、导致稀有类型丢失,过松则将大量技术噪声带入下游分析。
标准流程应包括可视化每项过滤前后分布、记录参数与样本对应信息,并结合污染校正和双细胞检测形成完整的高质量数据集。
标准 QC 流程总结表:
| 步骤 | 主要操作 | 建议阈值(需结合数据调整) |
|---|---|---|
| 细胞活性过滤 | nFeature / nCount | 200–500 < nFeature < 5000 |
| 线粒体比例过滤 | percent.mt | percent.mt < 10–20% |
| 基因表达过滤 | min.cells | min.cells > 3 |
| 环境 RNA 校正 | SoupX/DecontX | 按实际污染比例评估 |
| 双细胞检测 | DoubletFinder/scDblFinder/Scrublet | 默认工具建议 |
以上各项操作均已集成于主流分析框架的标准流程模板中,建议在质控环节后,仔细检查保留细胞和基因数量,并对异常样本进行追溯和重检,为后续下游分析打下坚实的质量基础。
Python质控(QC)流程模板
# -------------------------------------------------------------
# 单细胞 RNA-seq 质控全流程(Scanpy / scverse 生态)
# -------------------------------------------------------------
import scanpy as sc
import scrublet as scr # 双细胞检测
import matplotlib.pyplot as plt
from decontx import run as decontx_run # 环境 RNA 去除(可选)
# ---------- 0. 运行环境推荐 ----------
# conda create -n scQC python=3.11 scanpy scrublet decontx -c conda-forge
# conda activate scQC
# ---------- 1. 读入多批次 / 多样本 10x 原始计数 ----------
# *obs = 细胞 (barcode) × var = 基因*
sample_paths = {
"sample_A": "/path/to/sample_A/filtered_feature_bc_matrix/",
"sample_B": "/path/to/sample_B/filtered_feature_bc_matrix/",
}
adatas = []
for sname, spath in sample_paths.items():
ad = sc.read_10x_mtx(spath, var_names="gene_symbols")
ad.obs["sample"] = sname # 记录批次标签
adatas.append(ad)
adata = adatas[0].concatenate(*adatas[1:], batch_key="batch")
# ---------- 2. 计算基础 QC 指标 ----------
# n_genes_by_counts : 细胞内 *≥1 UMI* 的基因数 (nFeature)
# total_counts : 该细胞所有 UMI 总和 (nCount)
# pct_counts_mt : 线粒体 UMI 比例 (%) (percent.mt)
adata.var["mt"] = adata.var_names.str.upper().str.startswith("MT-")
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
# ---------- 3. 细胞过滤 ----------
# 原理:剔除低 RNA-content 细胞(裂膜/空滴)与极高 UMI 双细胞
adata = adata[
(adata.obs.n_genes_by_counts.between(300, 5_000)) & # nFeature
(adata.obs.pct_counts_mt < 10) # percent.mt
].copy()
# ---------- 4. 基因过滤 ----------
# 原理:至少在 min_cells 个细胞内被捕获的基因才保留
sc.pp.filter_genes(adata, min_cells=3)
# ---------- 5. 环境 RNA 去除(可选但推荐) ----------
# DecontX 使用贝叶斯分级模型估计“背景”UMI → 生成去污染矩阵
adata = decontx_run(adata, layer_raw="counts") # output layer: 'decontx_counts'
adata.X = adata.layers["decontx_counts"] # 替换为校正后表达矩阵
# ---------- 6. 双细胞 (doublet) 检测 ----------
# Scrublet 基于模拟混合表达模型,输出 doublet_score & 二分类预测
scrub = scr.Scrublet(adata.X)
dbl_scores, dbl_calls = scrub.scrub_doublets()
adata.obs["doublet_score"] = dbl_scores
adata.obs["predicted_doublet"] = dbl_calls
adata = adata[~adata.obs["predicted_doublet"], :] # 移除预测 doublet
# ---------- 7. 归一化 & 对数转换 ----------
# Normalization:将每细胞 UMI 归一到 1e4 → 计数规模可比
# Log1p:ln(UMI + 1),服从对数正态且稳定方差
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
# ---------- 8. 快速检查结果 ----------
sc.pl.violin(
adata,
keys=["n_genes_by_counts", "total_counts", "pct_counts_mt"],
jitter=0.4, multi_panel=True
)
print(adata)
R质控(QC)流程模板
###############################################################
## R / Seurat 单细胞 QC 流水线 + 双细胞检测 ##
## 依赖包:Seurat (>=5)、decontX(celda)、DoubletFinder、ggplot2、patchwork ##
###############################################################
# 0. 运行环境建议
# install.packages("Seurat")
# BiocManager::install("celda")
# remotes::install_github("chris-mcginnis-ucsf/DoubletFinder")
# install.packages(c("ggplot2", "patchwork"))
library(Seurat)
library(celda) # decontX()
library(DoubletFinder) # 双细胞检测
library(ggplot2)
library(patchwork)
# 1. 读取多个 10× mtx 目录并合并
sample_paths <- list(
SampleA = "/path/to/SampleA/filtered_feature_bc_matrix/",
SampleB = "/path/to/SampleB/filtered_feature_bc_matrix/",
SampleC = "/path/to/SampleC/filtered_feature_bc_matrix/"
)
seurat_list <- lapply(names(sample_paths), function(snm) {
mat <- Read10X(sample_paths[[snm]])
CreateSeuratObject(mat,
project = snm,
min.cells = 3,
min.features = 200)
})
sc <- merge(x = seurat_list[[1]],
y = seurat_list[-1],
add.cell.ids = names(sample_paths),
project = "scRNA")
# 2. 计算基础 QC 指标
sc[["percent.mt"]] <- PercentageFeatureSet(sc, pattern = "^MT-")
VlnPlot(sc, features = c("nFeature_RNA","nCount_RNA","percent.mt"))
FeatureScatter(sc, "nCount_RNA", "percent.mt") +
FeatureScatter(sc, "nCount_RNA", "nFeature_RNA")
# 3. 过滤低质量细胞
sc <- subset(
sc,
subset = nFeature_RNA > 300 &
nFeature_RNA < 5000 &
percent.mt < 10
)
# 4. 归一化、找高变基因、回归线粒体
sc <- NormalizeData(sc)
sc <- FindVariableFeatures(sc, nfeatures = 2000)
top10 <- head(VariableFeatures(sc), 10)
VariableFeaturePlot(sc) + LabelPoints(points = top10)
sc <- ScaleData(sc, vars.to.regress = "percent.mt")
# 5. 使用 decontX 校正 ambient RNA(每样本独立)
contam_vec <- vector("numeric", length = ncol(sc))
names(contam_vec) <- colnames(sc)
for (sid in names(sample_paths)) {
cell_idx <- which(sc$orig.ident == sid)
counts_mat <- GetAssayData(sc)[, cell_idx, drop = FALSE]
dc_res <- decontX(counts_mat)
contam_vec[cell_idx] <- dc_res$contamination
}
sc$contamination <- contam_vec
# 5.1 过滤高污染细胞
sc <- subset(sc, subset = contamination < 0.20)
# 6. 降维可视化
sc <- RunPCA(sc, features = VariableFeatures(sc))
sc <- RunUMAP(sc, dims = 1:30)
FeaturePlot(sc, features = "contamination") +
scale_color_viridis_c() +
ggtitle("UMAP — DecontX contamination")
# 7. 双细胞(doublet)检测分析(建议在 PCA/UMAP 后进行)
# 必须先 RunPCA/UMAP,且不应有过量低质/污染细胞
# 7.1 参数设置
nExp_poi <- round(ncol(sc) * 0.05) # 预估双细胞比例,通常2-8%,这里用5%经验值
sc <- FindNeighbors(sc, dims = 1:20)
sc <- FindClusters(sc, resolution = 0.5)
sc <- doubletFinder_v3(
sc,
PCs = 1:20,
pN = 0.25,
pK = 0.09, # 需参数扫描确定最优(详见官方 vignette)
nExp = nExp_poi
)
# 7.2 结果提取与过滤
doublet_col <- grep("^DF.classifications", colnames(sc@meta.data), value = TRUE)
table(sc[[doublet_col]][,1]) # 查看doublet/singlet分布
# 若要移除双细胞,仅保留 singlet
sc <- subset(sc, subset = get(doublet_col) == "Singlet")
# 7.3 可视化双细胞分布
DimPlot(sc, group.by = doublet_col, reduction = "umap") +
ggtitle("UMAP — DoubletFinder classifications")
# 8. 持久化保存
saveRDS(sc, file = "Seurat_QC_finished_doublet_filtered.rds")
🎯 本期 HighLight 精选
-
梳理了Seurat和Scanpy/AnnData的数据结构和底层逻辑,帮你轻松看懂R与Python生态的异同与优势 💾🔄
-
教会你QC关键指标的科学内涵,助你从容应对各种“过滤难题” 🧐
-
环境RNA校正、双细胞检测实用方法全收录,为你守住单细胞分析的底线 🧹🧪
-
R/Python互通技巧与标准模板一站式奉上,让切换和协作变得“丝滑” 🔁
-
强调数据可视化和参数灵活调整,让质控有据可依,结果一目了然 👀📊
📝 互动小测:你能突破 QC 小关卡吗?
1. 下列关于 Seurat 与 Scanpy/AnnData 对象的数据结构描述,哪一项是正确的?
A. Seurat 的表达矩阵默认行是细胞,列是基因
B. AnnData 的 obs 表存储的是基因注释信息
C. Seurat 的 meta.data 对应 AnnData 的 obs
D. AnnData 的 .X 属性不能存放稀疏矩阵
2. 某批单细胞数据中,部分细胞 nFeature < 100,线粒体比例正常。你应该如何处理?
A. 这些细胞大概率为低质量,应予以剔除
B. 保留并继续下游分析
C. 仅用线粒体比例作为过滤标准
D. 随机保留一部分
3. 如果你在环境 RNA 校正后发现部分基因的低表达在所有细胞中同步下降,最合理的解释是?
A. 这些基因主要来自环境污染,校正有效
B. 校正参数设置错误
C. 这些基因本身高表达
D. 数据本身没问题,属于正常现象
4. 当不同样本批次间的 QC 指标分布差异明显,你最优先的操作应该是?
A. 针对每个批次单独 QC,并对比过滤效果
B. 全部使用统一阈值,不区分批次
C. 只分析指标分布最好的批次
D. 忽略批次差异
💬 欢迎把你的答案和理由打在评论区!看看大家的操作姿势有没有“神同步”~
🚀 结束语
希望本期让你不再“谈 QC 色变”,敢于直面噪音与污染,把数据打磨得又纯又亮!有任何疑问、吐槽或独家秘籍,欢迎在评论区留言。
别忘了关注 EVBioX,下期带你搞定批次整合与下游分析,江湖再见!✨




















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











