




同一池子里捞到的细胞,其计数深浅差异如同光照明暗;归一化让你“调亮”画面,而特征提取决定你究竟看哪一块。
0. HighLight(90 s 速览)
-
📈 归一化四路流派+决策树:新增 SCTransform 2、pseudo-bulk/Atlas 场景与“不必高度归一化”的例外。
-
🔍 三大核心算法深拆:shifted-log、scran、Pearson residual——公式、踩坑点、R/Python 双语代码拆栏展示。
-
🎯 特征提取新范式:稀疏矩阵→维度灾难;在归一化框架内选 HVG 的最佳实践。
-
⚡ 速查备忘表:速度|内存|下游友好度一页比较。
1. 单细胞归一化:让每个细胞都有同样的“光照”
1.1 为什么要归一化?
还记得第一次打开原始单细胞表达矩阵时的震撼吗?同一类型的细胞,计数能差出几个数量级。这种差异,不全是生物学意义上的“基因表达”,更多时候是测序深度、捕获效率、甚至细胞本身大小等技术性噪声捣的乱。RNA 装得多“如果不做归一化,这些“假信号”会在下游分析时被错误地当成生物差异,结果就是“噪音大过信号”,看不清真实的细胞世界。
举个例子:你用10x平台测了20,000个细胞,每个细胞总reads从几千到几万。你会发现,不归一化直接用表达量聚类,最后分群结果大概率是“测得多的堆一起,测得少的堆一起”,和生物没半毛钱关系。所以哪怕是最简单的库量归一化,也能极大地减少这种假象。
1.2 归一化方法的四大家族
| 流派 | 核心思路 | 代表方法 | 通俗一句话 |
|---|---|---|---|
| Library-size scaling | 按每细胞总库量缩放 | CPM/TPM、TMM、RLE | “把盘子里先分成 1 万份再称重” |
| Variance-stabilizing transform | 让方差趋于常数 | √count、shifted-log、arcsinh | “开根号 / 加 log,抹平大数波动” |
| Model-based scaling | 拟合噪声模型求 size factor | scran、DESeq2、glmGamPoi | “和邻居组团砍价,求一个中位数折扣” |
| Residual / Latent | 直接用模型残差 | Pearson residual、SCTransform 2、scVI | “归一化+方差稳定一锅端” |
Side note 🤔
在 CITE-seq 的 ADT 或 ATAC 计数这类“天生低动态”数据上,最小化处理(total-count + log1p)反而最稳,不必大动干戈。
1.3 给你一棵“归一化决策树”
四个决策维度:细胞数 × 计算资源 × UMI/Smart-seq × 是否要跨批次整合
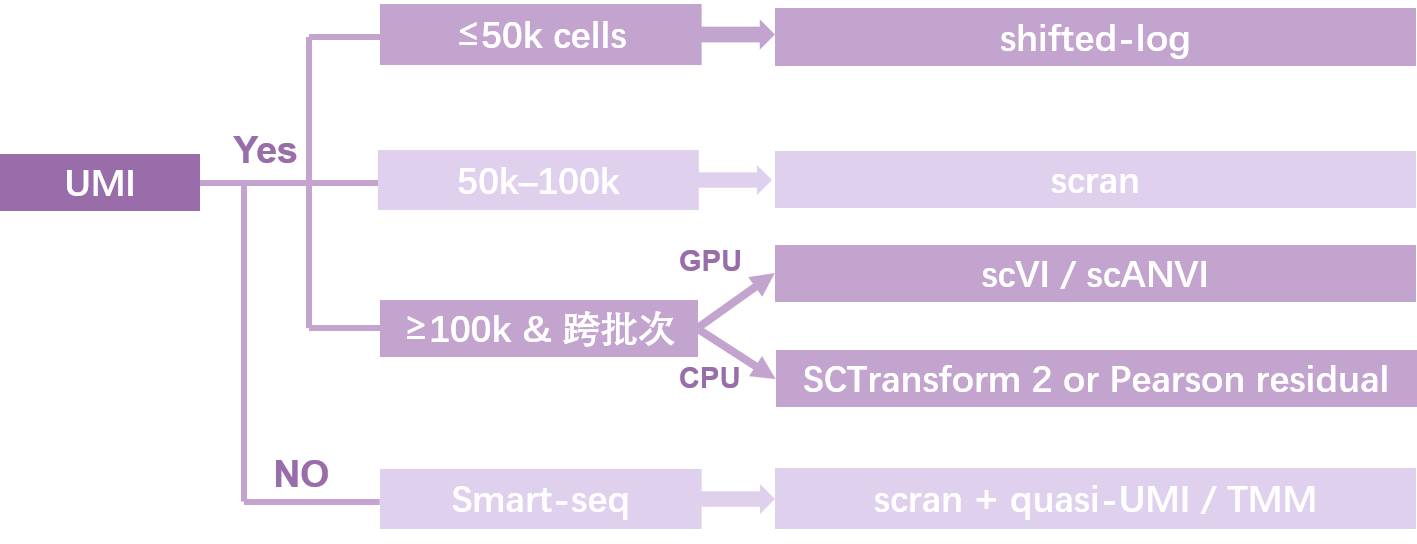
1.4 归一化常用方法横评表
为了便于临时查表对比,整理如下:
| 方法 | 速度 | 内存 | 下游分析友好度 | UMI适配 | non-UMI适配 | 场景举例 |
|---|---|---|---|---|---|---|
| Shifted-log | ★★★★★ | ★★☆☆☆ | 中等 | ✔ | △ | 10x常规质控、探索 |
| scran | ★★★☆☆ | ★★★☆☆ | 高 | ✔ | ✔ | 10x/Smart-seq2、批次差异较大 |
| SCTransform 2 | ★★☆☆☆ | ★★★★☆ | 极高 | ✔ | △ | 超大数据、批次整合 |
| Pearson residual | ★★☆☆☆ | ★★★★☆ | 极高 | ✔ | ✔ | 多批次/atlas级分析 |
2. 核心归一化方法拆解:从原理到代码
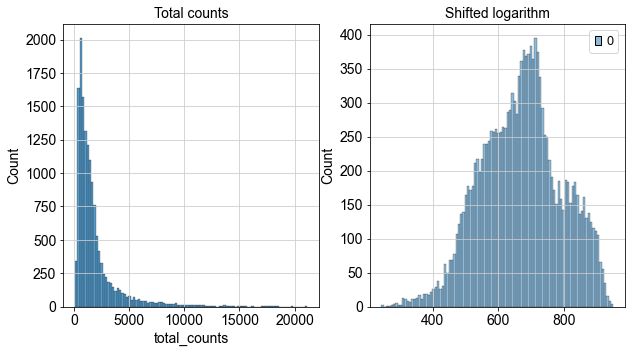
2.1 Shifted-log:最经典的“傻瓜”归一化
Shifted-log(加常数后的对数变换)本质就是每个细胞先按总量缩放(比如都换算成1万条reads),再对每个基因表达取log。这样做能让大多数基因都不会被极端高表达的细胞“拉爆”。
-
:原始计数(某基因在某细胞的表达量)。
-
:size factor,细胞的总表达量或测序深度的归一化因子,用于校正不同细胞的总计数差异。
-
:pseudo-count,假计数(如1),避免对0取对数带来的数学问题。
-
将原始计数先除以细胞总量标准化,再加上pseudo-count后取对数,从而压缩高表达值、拉伸低表达值,使不同细胞、不同基因间的表达量更易比较,同时减少测序深度等技术噪声对数据的影响。
-
:第c个细胞的size factor。
-
:基因g在细胞c的原始计数。
-
:细胞c所有基因的计数和(即该细胞的总表达量)。
-
:目标归一化总量(如10,000或1,000,000),常用中位数、1e4或1e6。
目的:计算每个细胞的归一化因子,将不同细胞的总表达量调整到统一的尺度,从而消除样本间测序深度或捕获效率的影响,是归一化的基础步骤。
pbmc <- NormalizeData(pbmc, normalization.method="LogNormalize", scale.factor=1e4)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
优点:速度极快,基本不挑数据类型。
缺点:如果测序深度悬殊太大,还是可能残留技术噪声,对低表达基因不够友好。
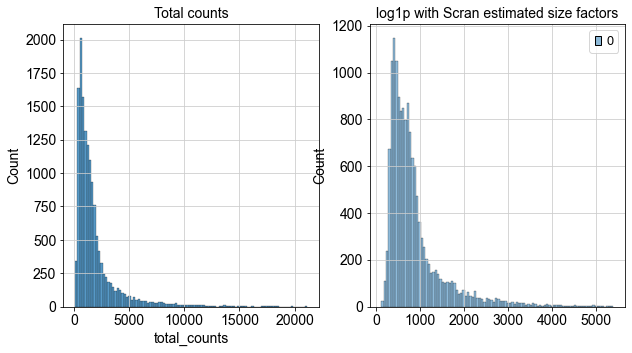
2.2 scran deconvolution:智能化的归一化新秀
scran最大的特色,是把细胞“组团”生成伪池,再根据各池子与全局的计数比例,推算每个细胞的size factor。换句话说,它会自动学习数据中“隐藏”的测序偏差。
sce <- computeSumFactors(sce)
sce <- logNormCounts(sce)
优点:对10x、Smart-seq2均适用,尤其适合深度/捕获率不均的数据。
缺点:细胞太少(<2k)或太多(>200k)时效率和精度都会受影响,需要先粗聚类分桶。
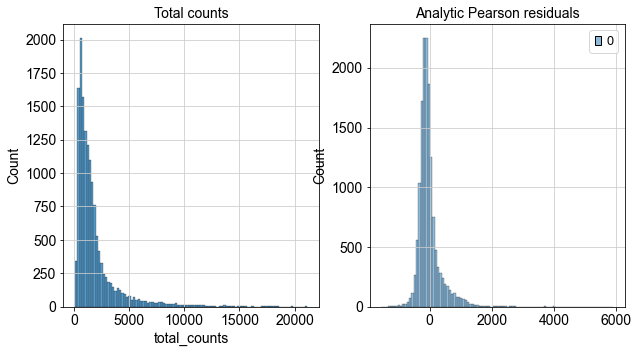
2.3 Pearson residual/SCTransform 2:一站式标准化神器
这套思路的核心是:先用负二项分布回归建模,预估每个基因在每个细胞的“应有”表达,再用实际值和期望值的残差作为新表达矩阵。这种方式天然把噪声和生物差异拆开,下游PCA/聚类不再受技术干扰。
SCTransform 2是目前Seurat的主推方案(特别是批次整合时),它对参数θ的估计更稳健,非UMI数据也能hold住。
pbmc <- SCTransform(pbmc, vst.flavor="v2")
sc.experimental.pp.normalize_pearson_residuals(adata, theta=100)
优点:对批次整合、复杂场景极其友好;输出数据已经方差稳定,下游可以直接用。
缺点:对算力有一定要求,大型项目推荐用多核/GPU。
3. 特征提取:帮你“找到故事最精彩的章节”
3.1 为什么要做特征提取?
单细胞测序给了我们动辄三万个基因、几万个细胞的“超高维”数据矩阵——但大部分基因,在大部分细胞里其实都是零。盲目全部输入后续分析,很容易让真正的信号被“背景噪声”稀释掉,甚至让内存爆炸。所以,我们要提前筛掉那些“无信息”基因,把降维、聚类的“舞台”留给最活跃、最有代表性的部分。
3.2 特征选择的原则与方法
不同归一化方法,对应着不同的高变基因(HVG)算法。选HVG的核心,就是找到在群体中变异最大、而又不是“高表背景噪声”的那一批。
| 类别 | 统计量 | 优点 | 缺点 | 典型场景 |
|---|---|---|---|---|
| 方差-均值 (vst) | 变异度 Z-score | 极快 | 深度敏感 | ≤ 50k 快速探索 |
| Deviance | NB/Poisson deviance | 抗深度、Smart-seq 友好 | 主要 R 实现 | Smart-seq2、non-UMI |
| Pearson-HVG | 残差方差 | 与 SCTransform 同根 | 需先算 residual | ≥100k、大批次 |
| Dropout-based | π₀ 模型 | 擅长捕捉“零主导”基因 | 忽视高表达长尾 | Capture 率偏低数据 |
实际操作中,Seurat 5的SCTransform 2高变基因直接在归一化流程中产出,极为便利。不同平台多批次时,建议每批先各自选一遍HVG,再求交集,以保留全局信号。
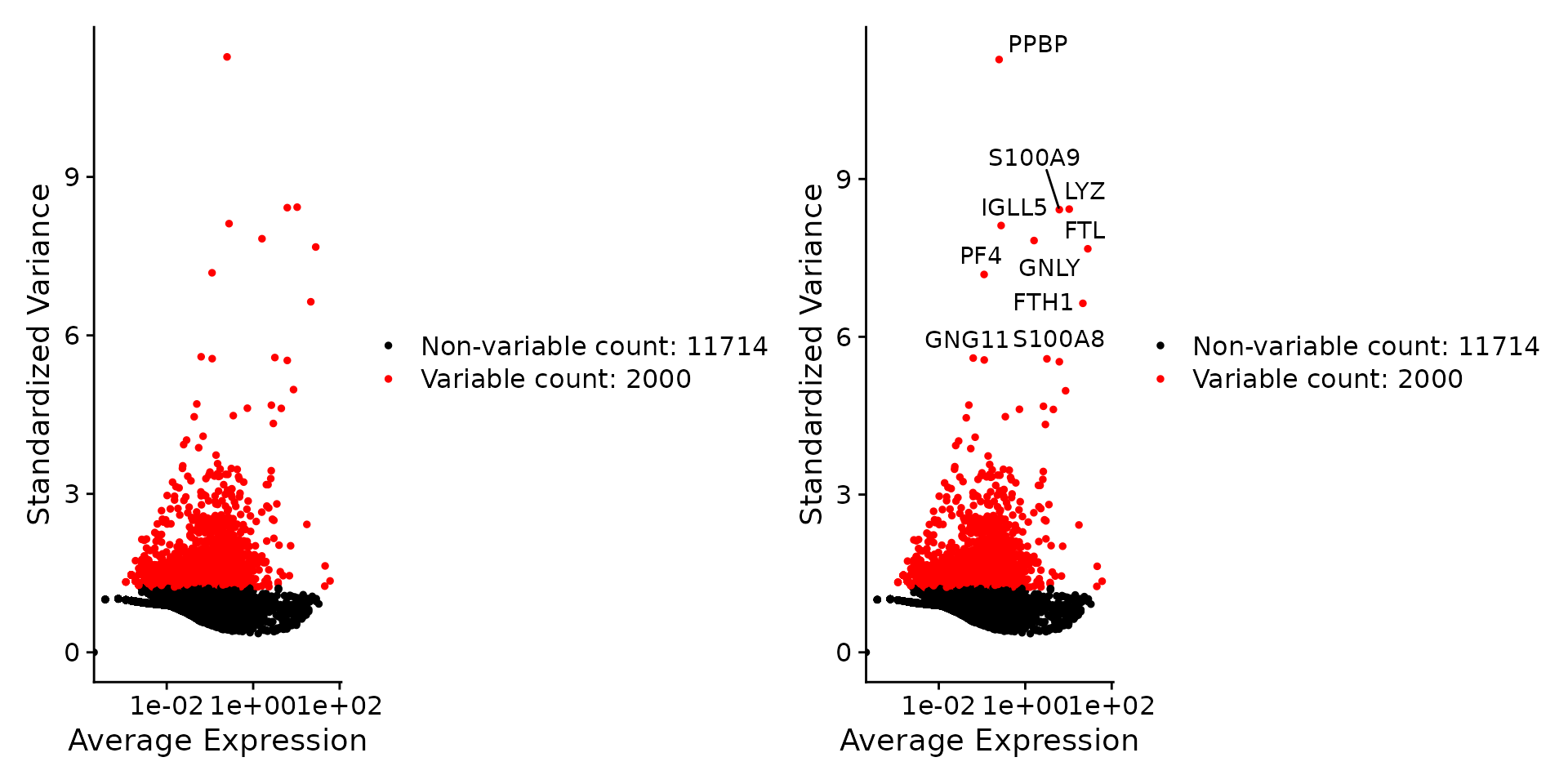
4. 不同场景下的归一化+特征提取“食谱”
| 场景 | 归一化 + HVG 组合 | 理由 |
|---|---|---|
| ≤ 50k | Shifted-log + HVG-dispersion | 易复现,训练课程常用 |
| ≥ 100k & 多批次 | scran ➞ Pearson-HVG 或 SCTransform 2 | 残差空间一致,整合稳 |
| Smart-seq2 | scran + deviance | 兼顾非 UMI 与 PCR 噪声 |
| 后续做 Trajectory / Batch-integration | Pearson residual / SCTransform 2 | 避免不同空间带来伪时序漂移 |
| CITE-seq / multiome | total-count → log1p + deviance | 动态范围窄,不必深度归一化 |
5. 实用Debug自查表(掉坑别慌,先排查)
| 问题 | 典型症状 | 快速排查 |
|---|---|---|
| size factor=0 | 全零细胞/空滴未过滤 | 先筛掉异常细胞,再归一化 |
| scran报“spikes not found” | ERCC标签缺失 | 加isSpike=FALSE参数 |
| Pearson θ爆表 | 深度过低导致 | 固定θ=100或选用SCTransform 2 |
| HVG数异常少 | ScaleData用早了 | 归一化后再选HVG,别提前缩放 |
| Pseudo-bulk/atlas多批次 | 批次极不均衡 | SCTransform 2+PrepSCTFindMarkers()组合 |
6. 归一化与特征提取——复用模板
Python模板
# 推荐在 conda 虚拟环境下安装
# conda install -c conda-forge scanpy anndata numpy pandas
# pip install anndata2ri # 若与R互操作
import scanpy as sc
import numpy as np
# ========== Step 1. 读取原始AnnData ==========
# adata = sc.read_10x_mtx("raw_matrix_path/")
# or
# adata = sc.read_h5ad("your_file.h5ad")
# ========== Step 2. 细胞/基因初步质控 ==========
# 过滤低质量细胞和低表达基因(具体阈值按需调整)
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
# ========== Step 3. 归一化方法选择 ==========
# 方法一:经典库量归一化 + log1p(10x平台/快速探索,适合≤50k细胞)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
# adata.raw = adata # 推荐存一份归一化数据用于后续差异分析
# # 方法二:Pearson残差归一化(推荐 ≥50k、批次整合/深度差异明显)
# sc.experimental.pp.normalize_pearson_residuals(adata, theta=100)
# # 方法三:sctransform风格(Scanpy兼容但多在R/Seurat用)
# # 推荐实际大项目时直接用R/Seurat的SCTransform,或通过anndata2ri接口
# ========== Step 4. 高可变基因(HVG)筛选 ==========
# 推荐与归一化方法配套
# 对log1p数据,经典方差法:
sc.pp.highly_variable_genes(
adata, flavor='seurat_v3', n_top_genes=2000, batch_key=None # 可加batch_key分批挑选
)
# # 对Pearson残差归一化后,建议用flavor="pearson_residuals"
# sc.pp.highly_variable_genes(
# adata, flavor="pearson_residuals", n_top_genes=2000
# )
# 可查看挑选结果
print(f"筛选到高变基因数: {np.sum(adata.var['highly_variable'])}")
# ========== Step 5. 下游分析准备 ==========
# 只保留高变基因进行后续降维/聚类
adata = adata[:, adata.var['highly_variable']].copy()
# 标准化(均值0方差1,供PCA/聚类使用,不影响差异分析)
sc.pp.scale(adata, max_value=10)
# ========== Step 6. 下游分析流程衔接 ==========
# 主成分分析
sc.tl.pca(adata, svd_solver='arpack')
# 邻接图与聚类
sc.pp.neighbors(adata, n_neighbors=15, n_pcs=40)
sc.tl.leiden(adata, resolution=0.5)
# 可视化
sc.tl.umap(adata)
sc.pl.umap(adata, color=['leiden'])
# 差异表达分析(可选,用原始/归一化数据)
# sc.tl.rank_genes_groups(adata, groupby='leiden', method='wilcoxon')
# sc.pl.rank_genes_groups(adata, n_genes=20, sharey=False)
✨ 代码注释和常用衔接Tips
-
adata.raw = adata:建议在归一化后存一份raw,用于后续marker/差异分析的参考。 -
若后续还要批次整合,建议归一化+HVG时加
batch_key=你的批次字段,保证不同批次挑到相似信号。 -
若用Pearson残差,后续PCA/聚类不需要再scale(其本身已方差标准化)。
-
大项目推荐批量输出每步QC和归一化后数据,便于回溯。
🚀 快速迁移到自己数据集
-
替换
adata = sc.read_10x_mtx(...)或read_h5ad(...)为你的文件路径。 -
调整质控、HVG筛选、归一化方法参数以适应项目实际需求。
-
下游可无缝衔接Scanpy主流程(降维、聚类、可视化、差异分析)。
R模板
# 推荐用renv管理依赖,或手动安装
# install.packages("Seurat")
# if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
# BiocManager::install(c("SingleCellExperiment", "scran"))
library(Seurat)
library(Matrix)
# library(SingleCellExperiment)
# library(scran) # 若用scran归一化
# ========== Step 1. 读取原始数据 ==========
# 一般10x数据:
# pbmc.data <- Read10X(data.dir = "filtered_gene_bc_matrices/hg19/")
# pbmc <- CreateSeuratObject(counts = pbmc.data, min.cells = 3, min.features = 200)
# ========== Step 2. 初步质控 ==========
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nCount_RNA > 500 & percent.mt < 10)
# percent.mt = PercentageFeatureSet(pbmc, pattern = "^MT-")
# ========== Step 3. 归一化选择 ==========
# 方法一:LogNormalize(Seurat默认,适合10x,≤50k细胞)
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
# # 方法二:SCTransform 2(推荐多批次/深度差异/大样本,自动归一+方差稳定+HVG)
# pbmc <- SCTransform(pbmc, vst.flavor = "v2", variable.features.n = 3000, verbose = FALSE)
# # 方法三:scran归一化(建议大批量或Smart-seq/非UMI/多技术整合场景)
# library(SingleCellExperiment)
# library(scran)
# sce <- as.SingleCellExperiment(pbmc)
# clusters <- quickCluster(sce)
# sce <- computeSumFactors(sce, clusters=clusters)
# sce <- logNormCounts(sce)
# pbmc[["scran"]] <- CreateAssayObject(counts = counts(sce), data = logcounts(sce))
# DefaultAssay(pbmc) <- "scran"
# ========== Step 4. 高可变基因(HVG)筛选 ==========
# 方法一配合LogNormalize/SCTransform:
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
# # SCTransform已自带HVG,无需重复筛选
# hvgs <- VariableFeatures(pbmc)
# ========== Step 5. 数据标准化(中心化/缩放,PCA聚类用,不影响差异分析) ==========
pbmc <- ScaleData(pbmc, features = hvgs) # 可设置max.cells.per.ident
# ========== Step 6. 下游分析流程衔接 ==========
pbmc <- RunPCA(pbmc, features = hvgs, npcs = 40, verbose = FALSE)
pbmc <- FindNeighbors(pbmc, dims = 1:20)
pbmc <- FindClusters(pbmc, resolution = 0.5)
pbmc <- RunUMAP(pbmc, dims = 1:20)
DimPlot(pbmc, reduction = "umap", group.by = "seurat_clusters")
# ========== Step 7. 差异表达分析/marker基因挖掘(用raw数据更科学) ==========
# pbmc <- SetAssayData(pbmc, slot = "data", new.data = pbmc@assays$RNA@counts)
# markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
💡 实用衔接Tips
-
SCTransform推荐用于大样本、批次整合、深度不均或后续需要Harmony/CCA等整合流程时。
-
若后续有批次整合/伪时序等需求,建议在归一化和选HVG时全流程指定同一批次字段。
-
scran方法适合Smart-seq2、非UMI或平台混合数据,可以显著提升深度不均的数据表现。
-
pbmc@assays$RNA@data和pbmc@assays$RNA@counts区别:前者是归一化表达量,后者是原始计数,差异分析建议用原始计数。
🚀 快速迁移到自己数据集
-
替换
Read10X和CreateSeuratObject等输入为你项目的原始路径和参数。 -
调整归一化和HVG选择方法及参数适应具体数据量与场景。
-
下游分析可直接接入Seurat标准流程。
7. 参考资料
-
[Townes et al., 2019, Genome Biology]Deviance HVG理论基础
-
Satija Lab官方文档SCTransform 2解读
-
scran官方文档与源代码
📋 你学“会”了吗?本期自测小练习
在正式进入下期预告前,欢迎你检验一下自己的掌握程度!下面几道题都有些“思考含量”,欢迎把你的答案和理由写在评论区和大家一起讨论👇:
1. 单细胞测序数据A,UMI平台、包含8万个细胞、来自多个批次,每个批次的测序深度差异很大。如果你想后续做批次整合和细胞伪时序分析,下面哪种归一化+高变基因筛选的组合更合适?
A. CPM归一化 + 方差选HVG
B. scran归一化 + deviance选HVG
C. SCTransform 2归一化 + Pearson-HVG
D. 仅做库量归一化,不选HVG
2. 某研究者用Smart-seq2平台测了2000个细胞,捕获率和PCR效率差异很大,部分基因在大部分细胞为0,极少数细胞中高表达。如果想保证下游聚类和差异分析更准确,归一化和特征选择的推荐做法是?
A. Shifted-log归一化 + 方差HVG
B. scran归一化 + deviance-based HVG
C. Pearson residual归一化 + Dropout-based HVG
D. CPM归一化 + 随机选择部分基因
3. 你拿到一个CITE-seq(RNA+ADT)数据集,细胞总量12万,RNA部分测序深度高,ADT部分信号本身动态范围很窄。关于归一化,下列最合适的描述是?
A. RNA和ADT都用SCTransform 2归一化
B. RNA用shifted-log归一化,ADT只做最基础的库量归一化+log1p
C. RNA和ADT都做scran归一化
D. 只对ADT部分做方差稳定变换
4. 有一批样本,预处理时直接选了高变基因(HVG),后面才发现部分细胞测序深度远低于预期。请问这会造成什么潜在影响?
A. HVG名单可能被技术噪声主导,丢失生物信号
B. 高变基因筛选结果更准确
C. 归一化前选HVG无影响
D. 深度低反而能突出稀有细胞
你的答案和理由是什么?欢迎写在评论区,我们一起交流!(下期我会挑出优秀解答再做点评 😎)
🔮 下期预告
批次整合与细胞图谱映射篇,全面对比Harmony、BBKNN、scANVI等主流工具,带你体验“细胞世界的无缝拼接术”。敬请期待!
如有疑问、想法或想晒分数,评论区等你来战!
让EVBioX成为你科学思考、技术成长的小宇宙。



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











