



 本文介绍了一种不使用爬虫技术,仅通过修改网页源码来获取CellType基因数据的方法。作者通过调整表格显示记录数,成功下载了25000条基因与细胞类型对应的数据。
本文介绍了一种不使用爬虫技术,仅通过修改网页源码来获取CellType基因数据的方法。作者通过调整表格显示记录数,成功下载了25000条基因与细胞类型对应的数据。
我想下载每种细胞对应的marker gene。
1. 通过观察这个页面 Cell Type | Single Cell Atlas
发现基因和cell type的对应表格。但是怎么下载呢?
2. 通过观察代码,发现每次对表格翻页的时候,实际上调用了 Cell Type Profile
这个页面。看来网站使用shinyapp搭的。从shinyapp的口子进去,再看源码。
3. 发现在表格显示多少行记录那里的源码可以修改
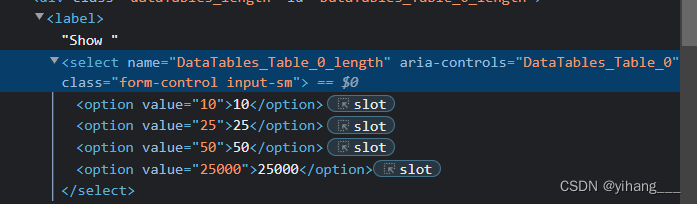
我直接把100改成了25000 结果进去后真的加载了25000行。。。于是复制粘贴就把数据弄下来了。
以上,完全没有用到爬虫。


















 648
648

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









