







SPMM 是 Structure-Property Multi-Modal foundation model 的简称,一种多模态分子性质-结构双向预训练模型。作者是韩国科学技术研究院人工智能研究生院,大田,韩国的 Jong Chul Ye,于 2024 年 3 月 14 日 发表在 nature communication 期刊上。
一、背景介绍
SPMM 是 Structure-Property Multi-Modal foundation model 的简称,来源于韩国科学技术研究院人工智能研究生院的 Jong Chul Ye 为通讯作者的文章:《Bidirectional generation of structure and properties through a single molecular foundation model》。文章链接:https://www.nature.com/articles/s41467-024-46440-3。该文章于 2024 年 3 月 14 日发表在 《nature communications》上。

近年来人工智能领域基础模型的成功催生了化学预训练模型的出现。这些模型为下游任务提供了有用的表示,但在分子领域进行多模态预训练的方法仍然有限。为了解决这个问题,作者提出了一种多模态分子预训练模型 SPMM,结合了结构和生化性质的模态。提出的数据处理和训练目标的模型管道将结构/性质特征对齐于一个共同的嵌入空间,使模型能够考虑分子结构和性质之间的双向信息。这些贡献产生了协同知识,能够通过单一模型处理多模态和单模态下游任务。通过一系列的实验,证明了 SPMM 能够解决各种有意义的化学挑战,包括条件分子生成、性质预测、分子分类和反应预测。
二、模型介绍
2.1 模型框架
捕获化合物及其性质之间的复杂关系是众多化学挑战的核心问题。受到计算机视觉领域的多模态学习模型的成功启发,近来的许多研究试图通过利用不同数据表示的知识来获得分子的更好特征。但这些工作仅引入了多模态以增强分子特征,而非用于这些不同模态之间的相互作用。并且由于 SMILES、InChI 和图表示几乎包含了分子中原子连接的相同信息,因此通过这些不同分子表示进行多模态学习不太可能带来新的性质。
在本研究中,作者关注分子结构和相关性质之间的跨模态理解,这有助于解决许多应用中的重要任务,如性质预测、条件分子设计等。这不同于多任务学习方法,后者将准备好的性质作为标签来提取通用特征,而作者将一组性质视为独立的模态,表示输入分子,并提出通过这种性质模态进行多模态学习可以提供更加有意义的特征。具体来说,作者提出了分子结构-性质多模态基础模型(SPMM),该模型允许通过广泛的分子结构和其性质向量进行计算化学实验。通过采用 Transformer 架构,该模型可以分别通过自注意力和交叉注意力机制完成模态内特征提取和模态间融合。
本研究的试验结果表明,通过单一基础模型同时学习结构特征与相关性质信息可以为各种下游任务提供更好的表示。具体而言,通过对结构和性质的对称处理,该模型可以实现双向生成和预测,这是以前无法做到的。
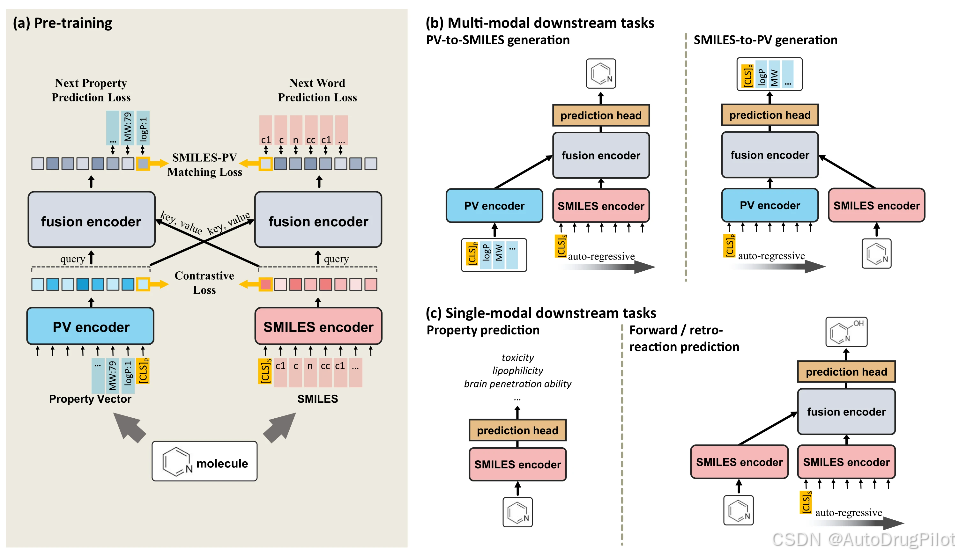
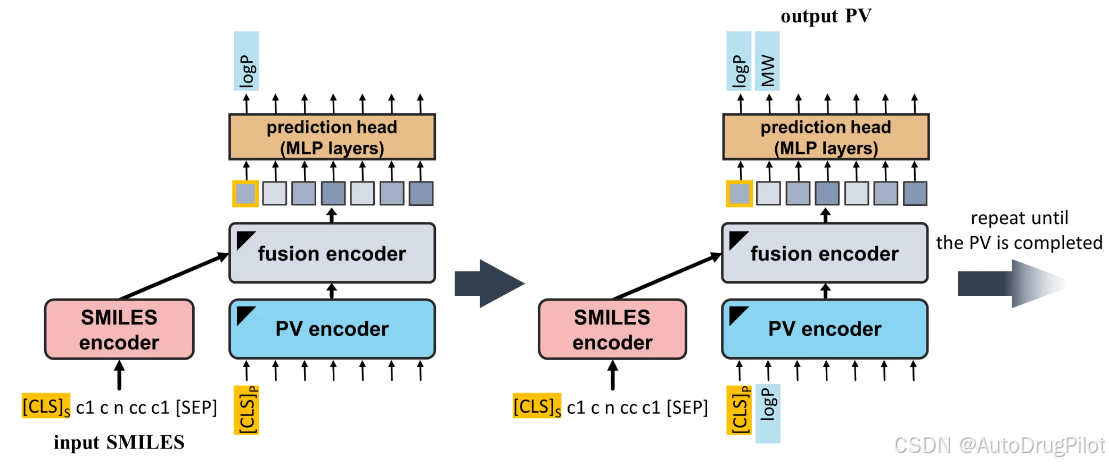
下图 a 展示了 SPMM 的整体模型架构和训练目标。SPMM 的框架扩展了双流 VLP 模型的结构,为每个模态输入编码一个单模态编码器,然后使用另一个编码器模块通过使用一种模态特征作为查询,另一种模态特征作为键/值来执行交叉注意。当给定训练分子时,SPMM 将分子的 SMILES 字符串及其性质向量(PV)作为多模态数据输入。SMILES 和 PV 通过各自的单模态编码器,执行自注意力,将嵌入的输入作为键、查询和值。在获得两个单模态特征后,对比学习将 SMILES 和 PV 特征对齐到相同的嵌入空间,这已被证明可以通过使跨模态编码更容易来提高模型性能,并引导单模态编码特征反映输入的更多语义。然后,编码后的 SMILES 和 PV 特征通过融合编码器,该编码器在 SMILES 和 PV 特征之间执行交叉注意。由于对比学习将 SMILES 编码器和 PV 编码器的输出对齐到相同的特征空间,单个融合编码器可以通过交替查询和键/值输入执行交叉注意。融合编码器通过 SMILES 的下一个词预测(NWP)、下一个性质预测(NPP)和 SMILES-PV 匹配损失(SPM)进行预训练。从给定的 Transformer 输入预测下一个组成部分是一种常用的自监督学习目标,NWP 和 NPP 任务使模型在另一种模态的语义特征的帮助下学习 SMILES 标记和性质之间的上下文关系。此外,SPM 预测给定的 SMILES 和 PV 对是否代表相同的分子。
一旦训练完成,SPMM 可以用于各种需要理解 SMILES 和性质的双向下游任务,如性质预测(从 SMILES 到性质)和性质条件下的分子生成(从性质到 SMILES,也称为反向 QSAR ),如下图 b 所示。此外,使用的预训练目标还允许预训练的 SPMM 用于单模态任务,如分子分类和反应预测(见下图 c)。预训练的 SPMM 在这些单模态任务中显示了与最新模型相当的性能,表明该模型作为基础模型的泛化能力。
2.2 数据准备
作者从 PubChem 获取了 50,000,000 个通用分子的 SMILES 用于预训练。使用的 53 种性质都可以通过 RDKit Python 库利用 SMILES 进行计算。用于 MoleculeNet 下游任务的数据集由 DeepChem Python 库提供。使用 DeepChem 的 scaffold splitter 对每个数据集进行 8:1:1 的训练/验证/测试集划分,这相比随机划分为模型提供了更严苛的条件。对于反应预测任务,使用了 USPTO-480k 数据集,该数据集包含 479,035 对反应物及其反应的主要产物。逆反应预测任务使用了 USPTO-50k 数据集,包含 50,037 对产物-反应物对及其对应的反应类型。尽管 USPTO-50k 数据集为每条反应数据提供了反应类型标签,但作者没有使用它们。
2.3 模型性能
2.3.1 模型学习了 SMILES 与性质之间的双向理解
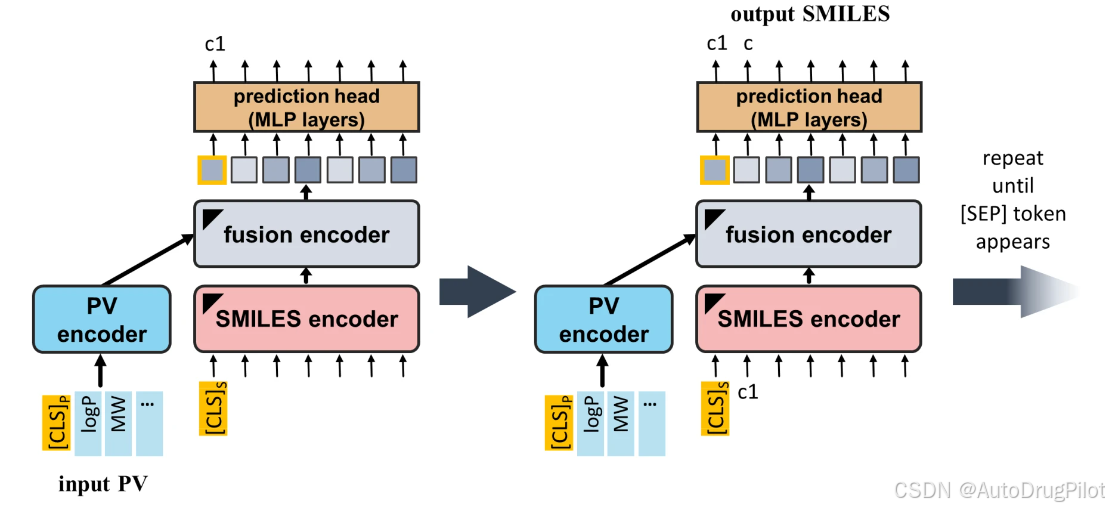
一旦 SPMM 预训练完成,就可以仅通过给定的 PV(性质向量)输入让模型生成 SMILES,这是许多化学任务(如新药分子设计)的关键挑战。为了展示 SPMM 的分子生成能力,作者准备了多种 PV 到 SMILES 的生成场景,并让预训练好的 SPMM 使用输入的性质自回归地生成 SMILES。这一过程与序列到序列的翻译任务非常相似(如下图所示),从 PV 的“性质句子”到 SMILES 的“分子结构句子”。
生成分子的有效性、唯一性和新颖性是 SPMM 分子生成的量化指标。此外,作为定性指标,测量了生成的 SMILES 与输入性质的匹配度,使用的是输入条件与生成分子性质之间的标准化均方根误差(normalized RMSE)。更具体地说,计算了所有控制性质的 RMSE 平均值,并将这些值根据预训练数据集中的每个性质的平均值和标准偏差进行了标准化处理。需要注意的是,RMSE 是在每个性质的标准化尺度上计算的,因为这些性质的值跨越了多个数量级。
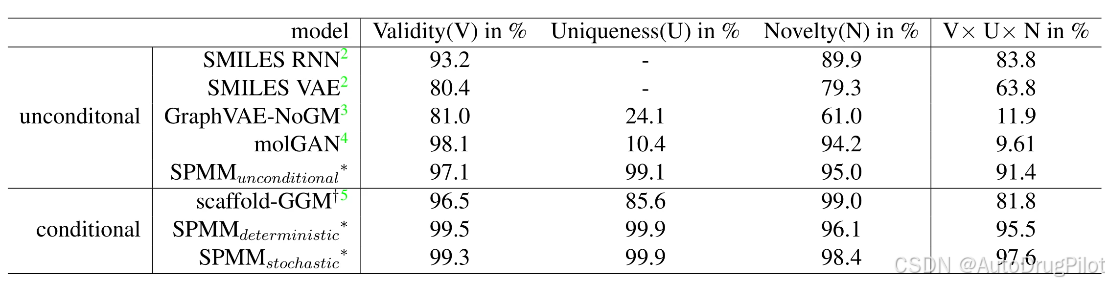
对于第一个 PV 到 SMILES 的生成场景,准备了来自 PubChem 的 1000 个 SMILES 的 PV,这些数据集不包含在预训练数据集中,并将它们输入预训练好的 SPMM 以生成相应的 SMILES。在此过程中,采样是以确定性方式完成的:从 SMILES 的 [CLS] 标记 ([CLS] S) 开始,模型预测下一个标记的概率分布,并选择概率最高的选项。下表的第一行展示了其结果。在 1000 个 PV 的确定性 PV 到 SMILES 生成输出中,99.5% 的生成输出是有效的 SMILES。53 种标准化性质的平均 RMSE 为 0.216,这表明生成样本的性质与输入性质相符。
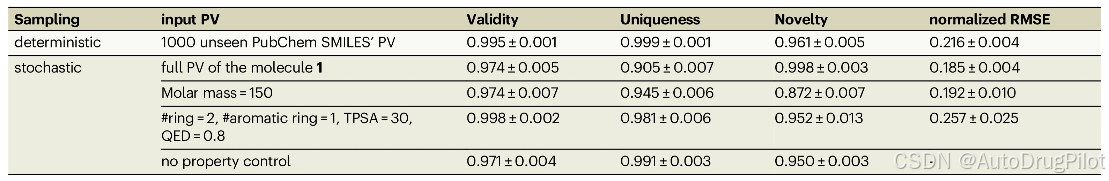
药物发现等应用领域通常要求为单一目标性质条件生成多个分子。为此,可以通过从建模的概率分布中随机采样下一个标记,而不是使用概率最高的标记。为了验证模型从单个 PV 输入生成多个分子的能力,作者使用固定的 PV 进行随机采样生成了 1000 个 SMILES。
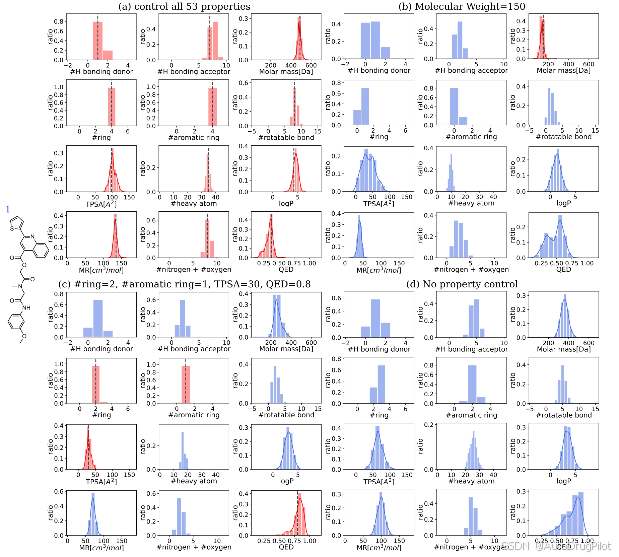
下图显示了从单个 PV 输入生成的 1000 个分子的性质分布。每个性质分布的众数落在输入的性质值上(图 a)。在仅给定部分性质的情况下,模型只关注已知的性质,而其他被遮蔽的性质不受限制(图 b、c)。SPMM 甚至可以在没有任何性质信息的情况下生成分子;当所有输入性质都被 [UNK] 标记替换时(图 d),模型进行无条件分子生成,输出遵循预训练数据集的分布。上表中的“stochastic”行列出了下图中条件下生成分子的有效性、唯一性和新颖性。根据性质输入的可行性或难度,这些指标有所波动,但在大多数情况下,它们都大于 0.9 。
如下表所示,在无条件和有条件分子生成场景中,与其他基准模型相比,SPMM 在生成有效、新颖和所需分子方面表现更好。
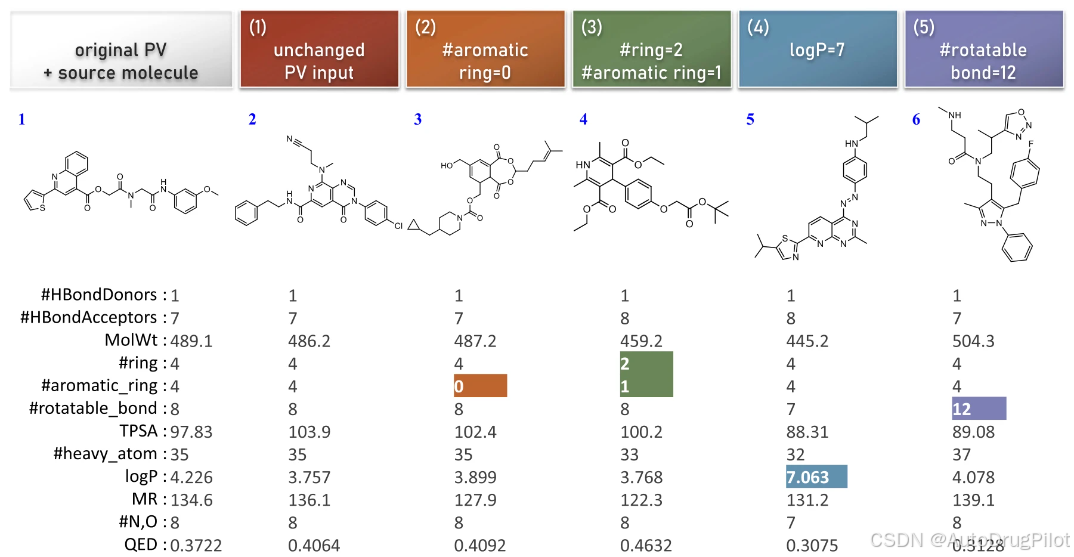
上述结果表明,SPMM 可以通过任意 PV 输入进行分子生成,从而实现简单的分子设计和编辑。作为分子编辑的可能示例,下图显示了 SPMM 对五个 PV 输入进行随机分子生成的输出,这些 PV 都源自同一分子的 PV,但其中四个值发生了变化。生成的分子遵循输入的修改,同时保持未修改的性质相似。SPMM 甚至能够生成具有域外条件的分子,例如“log P=7”(约 5% 的预训练数据集中 log P 大于 7)。
关于 SPMM 的整体分子生成性能,作者强调的是,SPMM 能够为模型在预训练中未见过的许多性质条件生成合适的 SMILES。当我们在训练 SPMM 时没有使用 50% 的随机性质遮蔽(即 [UNK] 标记),模型仅在给定全部 53 种性质时才有效,因为模型未见过部分给定的性质。然而,即便采用 [UNK] 标记遮蔽技术,模型在预训练过程中也无法面对大多数 2^53 种可能的性质组合。SPMM 处理任意性质条件生成 SMILES 的能力源于将 PV 视为“包含 53 个单词的语言”,并专注于每种性质的独立处理,而不是简单地将整个性质输入视为单一条件。这种创新的条件分子生成方法是现有方法中从未展示过的,因此可以用于许多重要的化学领域。
与 SMILES 生成采用相同的方法,预训练的 SPMM 还可以通过仅给定 SMILES 输入来生成 PV。这个任务等价于同时对给定的 SMILES 进行 53 种性质的预测。与 PV 到 SMILES 生成类似,性质是以自回归方式预测的:模型仅使用性质 [CLS] 标记 ([CLS] P) 预测第一个性质值,然后再次采用所有先前的输出以得到下一个预测值,依此类推(见下图 )。虽然使用的 53 种性质可以通过 Python 模块计算得到,但该实验的目的是验证基于数据驱动的性质估算与分析方法一致。
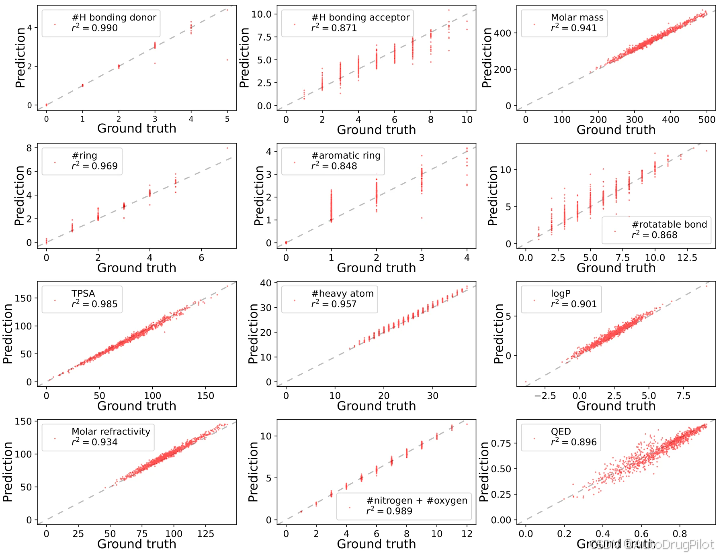
作者将 1000 个来自 ZINC15 数据集的 SMILES(这些数据不包含在预训练数据集中)输入预训练好的 SPMM,生成相应的 PV。下图是用于预训练的 53 种性质中选出的 12 种性质的真实值与生成输出的散点图。显然,SPMM 预测的性质非常接近实际值,大多数数据点都位于 y=x 线上。尽管模型由于 50% 的随机性质遮蔽几乎从未见过完整的 PV,但它能够自回归地预测所有 53 种性质。53 种性质的平均 r² 得分为 0.924。
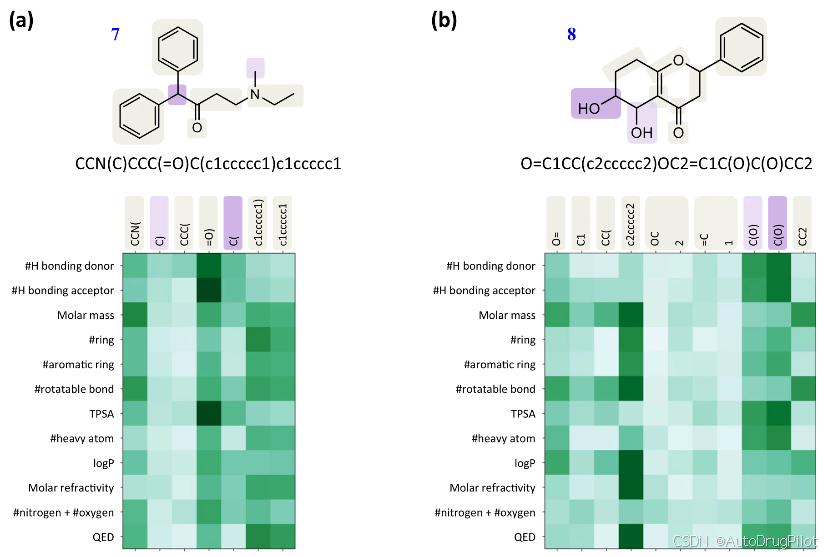
为了进一步解释预训练的 SPMM 所表现出的性能,作者通过可视化预训练 SPMM 的注意力得分分析了其学到的 SMILES 与性质向量之间的跨模态理解。基于 Transformer 的模型具有直观的注意力可视化的优势,能够展示模型如何通过提供输入查询与键之间的交叉注意力得分来考虑它们之间的关系。如下图所示,作者绘制了当 SMILES 和其性质向量输入时,预训练 SPMM 最后融合层的交叉注意力得分。由于交叉注意力有多个头,所以取了它们注意力得分的平均值。交叉注意力得分的模式与化学性质和分子片段之间的直观关系相吻合。与氢键形成相关的性质(如“NumHDonors”、“NumHAcceptors”)在含氧和氮原子的标记上表现出较高的注意力得分。性质“RingCount”主要集中在涉及环的标记上,而对侧链基团的注意力较弱,而性质“NumAromaticRings”仅对芳环成分给予较高的注意力得分。当不同的 SMILES 标记在分子中发挥类似作用时(如分子 7 中的“c1ccccc1)”和“c1ccccc1”),它们的注意力模式也类似。这个结果表明,SPMM 能够在没有显式监督的情况下捕捉分子结构与化学性质之间的关系。为了进行更深入的统计分析,作者还观察了哪些标记在 12 个选定的性质中显示出高注意力得分,使用了 1000 个随机采样分子的交叉注意力图。结果显示,与某一特定性质频繁相关的标记往往对该性质表现出高注意力得分;例如,“TPSA”在涉及极性原子的标记(如氧和卤素原子)上得分较高,“NumHAcceptors”与氢键形成相关的标记得分较高,而“NumAromaticRings”则与芳环成分相关。
2.3.2 作为分子基础模型的泛化能力
至此,作者已经证明了预训练的 SPMM 可以应用于需要理解 SMILES 与性质之间关系的任务。双流 VLP 模型结构的一个优势在于,SPMM 的多模态预训练过程包括调整一个单模态编码器的输出,通过与另一个单模态编码器的输出对齐以包含来自其他模态的上下文信息。这意味着 SMILES 编码器的输出不仅嵌入了输入分子的结构信息,还增强了其性质信息。
作者接着分析了预训练模型是否学习到了可以用于其他任务的信息表示,仅使用预训练 SPMM 的 SMILES 编码器(如下图),并对九个 MoleculeNet 下游任务和一个药物诱导肝损伤(DILI)预测任务进行了基准研究。每个 MoleculeNet 任务都是用于制药/生化应用的回归或分类任务,如溶解度、毒性和脑穿透性。DILI 分类任务旨在克服开放数据库的潜在限制,并验证 SPMM 是否可以扩展到更复杂的终点。该任务是分类给定分子是否有引起肝损伤的风险。
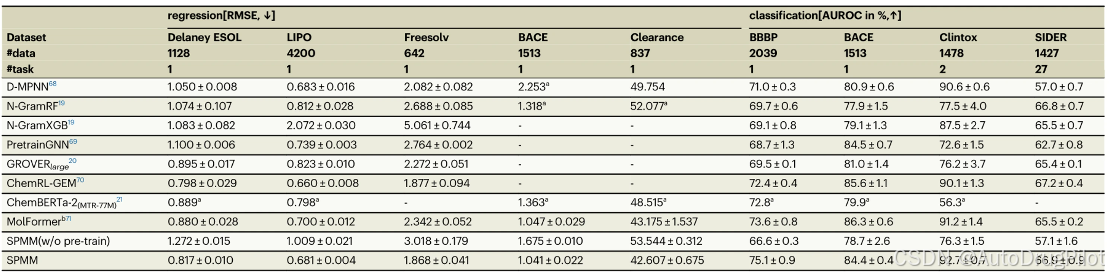
下表列出了 SPMM 和其他模型在 MoleculeNet 上的性能。仅使用 6 层 BERT 编码器,SPMM 在所有任务上显示出与最先进模型相当的性能。在九个任务中,SPMM 在五个任务上达到了最佳性能,展现了其作为基础模型的能力。我们还观察到,在没有预训练的情况下,模型的得分显著下降。
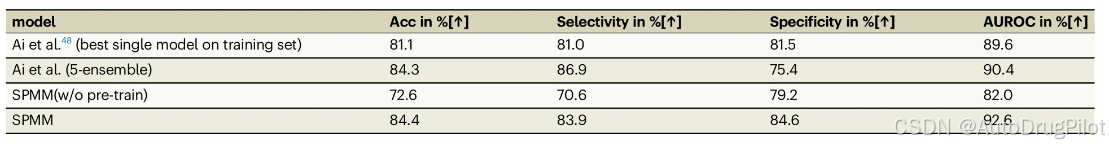
如下表所示,SPMM 在 DILI 分类任务中也优于提出的 5 个集成模型,而没有经过预训练的简单 BERT 层则不具备这种性能。
作者还对 SPMM 进行了正向和逆反应预测任务的训练,这要求模型从反应物 SMILES 预测产物 SMILES,反之亦然。将这两个任务视为序列到序列生成,这些反应预测任务的模型管道与 PV 到 SMILES 生成任务相同,只是将 PV 编码器替换为 SMILES 编码器(如下图)。
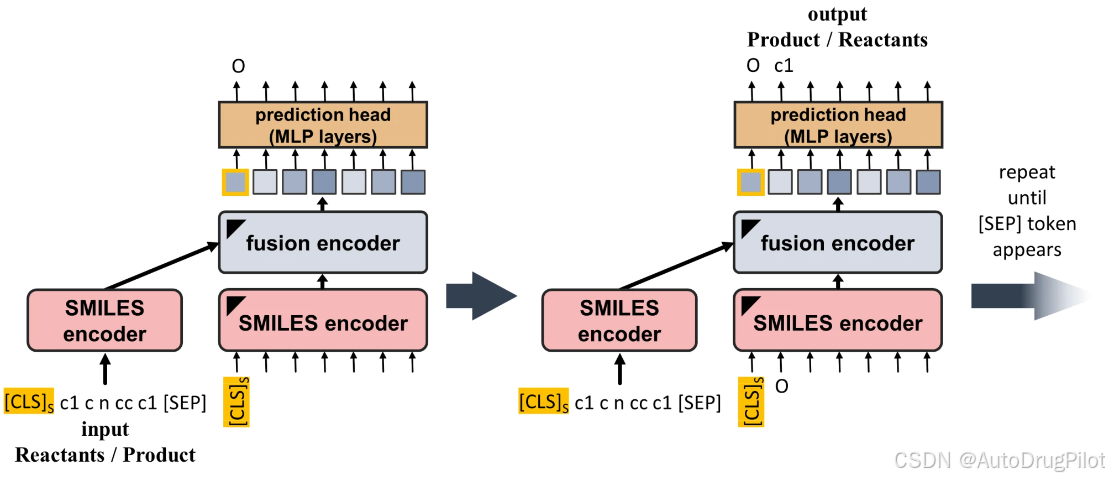
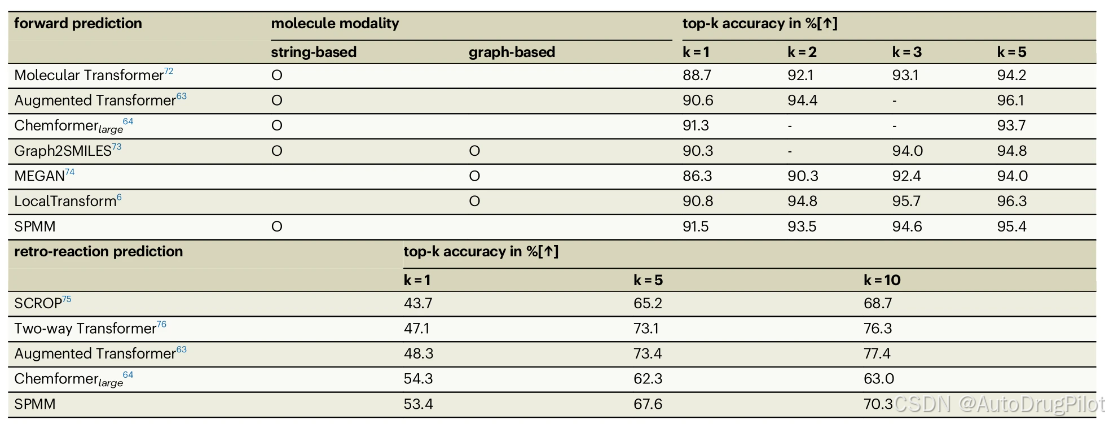
下表显示了 SPMM 和其他基准模型在正向和逆反应预测任务上的性能。尽管反应预测任务并不是性质涌现特征发挥显著作用的最佳场景,但 SPMM 在正向反应任务中以相对较小的预训练数据规模(即 5000 万个分子,相比于 Chemformer 的 1 亿个分子)实现了最高的 top-1 准确率。在基于字符串的逆反应任务模型中,SPMM 也获得了第二好的 top-1 准确率。
三、SPMM 评测
3.1 安装环境
复制代码项目:
git clone https://github.com/jinhojsk515/spmm.git根据项目提供的 requirements.txt 创建 SPMM 环境
conda create -n SPMM python=3.10
conda activate SPMM
pip install -r requirements.txt项目提供训练好的模型和数据集,保存在谷歌网盘,链接为:https://drive.google.com/drive/folders/1ARrSg9kXdXAL5VGgDBwizpSgcJwauPua ,如下所示。训练好的模型 checkpoint_SPMM.ckpt 保存在 ./pretrain 文件夹中。
网盘文件下载后放在项目目录下,完整文件目录情况如下:
.
|-- LICENSE
|-- README.md
|-- SPMM_models.py
|-- SPMM_models_rxn.py
|-- SPMM_pretrain.py
|-- calc_property.py
|-- config_bert.json
|-- config_bert_property.json
|-- config_bert_smiles.json
|-- d_classification.py
|-- d_classification_multilabel.py
|-- d_pv2smiles_batched.py
|-- d_pv2smiles_single.py
|-- d_regression.py
|-- d_rxn_prediction.py
|-- d_smiles2pv.py
|-- data
|-- dataset.py
|-- normalize.pkl
|-- p2s_input.csv
|-- pretrain
|-- property_name.txt
|-- requirements.txt
|-- s2p_input.txt
|-- scheduler
|-- vocab_bpe_300.txt
`-- xbert.py
3 directories, 24 files
其中,./data 文件夹中包含项目实验所需的数据。
./pretrain 文件夹中包含预训练好的 SPMM 模型 checkpoint_SPMM.ckpt 。
vocab_bpe_300.txt 包含用于 SMILES 分词器(tokenizer)所需的 token 。property_name.txt 中包含 NumHAcceptors(氢键受体)、NumHDonors(氢键供体)和 QED(药物定量估计)等 53 个化学属性的名字。
normalize.pkl 包含项目用于 PV 的 53 个化学属性的均值和标准差。
calc_property.py 用于计算化学属性,针对给定的 SMILES 建立一个 PV 。相应地修改此代码,可以将 SPMM 预训练用于自定义 PV。
SPMM_models.py 中包含 SPMM 模型和预训练的代码。
SPMM_pretrain.py 是 SPMM 进行预训练的脚本。
d_*.py 都是一些下游任务的脚本。
3.2 PV 到 SMILES 的生成
项目提供脚本 d_pv2smiles_single.py,可以根据输入的分子属性(PV,property vector)直接生成分子的 SMILES ,输入的分子属性以表格的形式输入,项目提供的示例表格是 ./p2s_input.csv,其中的属性设置如下。如果有其他的属性要求,直接在该表格中添加模型允许的属性和对应的数值即可。
property,input_value
QED,0.8
TPSA,30
RingCount,2
NumAromaticRings,1我们根据输入的属性要求生成分子,使用项目提供的训练好的模型 ./Pretrain/checkpoint_SPMM.ckpt ,测试生成 100 个分子,每次使用不同的随机种子生成分子。具体的命令如下:
python d_pv2smiles_single.py \
--checkpoint './Pretrain/checkpoint_SPMM.ckpt' \
--n_generate 100 \
--stochastic True \
--k 2生成过程输出如下:
seed: 689 True
Creating model
LOADING PRETRAINED MODEL..
load checkpoint from ./Pretrain/checkpoint_SPMM.ckpt
_IncompatibleKeys(missing_keys=['property_encoder.embeddings.word_embeddings.weight', 'property_encoder_m.embeddings.word_embeddings.weight'], unexpected_keys=['temp'])
==================================================
PV-to-SMILES generation in stochastic manner with k=2...
100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 100/100 [00:30<00:00, 3.32it/s]
mean of controlled properties' normalized RMSE: 0.26725107431411743
validity: 1.0
uniqueness: 1.0
Generated molecules are saved in 'generated_molecules.txt'生成过程占用显存 2 GB,用时大约 1 分钟。设置生成 100 个分子,实际生成 100 个分子并保存在 ./generated_molecules.txt 中,所有生成分子通过分子有效性和唯一性检查,部分生成分子如下所示:
COc1cc(C)c(OCC2CCCCO2)c(C(C)(C)C)c1
CN1CCC(CCNC(=O)c2cc(C(F)(F)F)ccc2F)C1
CCN1CCC(CCNC(=O)c2ccc(C(F)(F)F)c(Cl)c2)CC1
CCN1CCC(N(C)Cc2ccccc2C(F)(F)F)CC1C(=O)NC1CC1
CC(C(=O)NC1CCCCC1)c1ccc(C(F)(F)F)cc1Cl
...对应的分子结构如下所示。分子结构符合 QED 为 0.8,TPSA 30,环数为2,芳香环数量为 1 的需求。

3.3 SMILES 到 PV 的检查
项目提供脚本 d_smiles2pv.py,可以根据输入的分子 SMILES 生成分子属性(PV,property vector) 。项目提供训练集的 53 个属性均值和标准差文件 normalize.pkl 。脚本输入分子的 SMILES ,检查这些分子和训练数据集的属性差异情况。使用项目训练好的模型 ./Pretrain/checkpoint_SPMM.ckpt ,输入是从 pubchem 中挑选出不在训练集中的 1000 个模型未见过的分子 SMILES ,即 ./data/2_PV2SMILES/pubchem_1k_unseen.txt 。具体命令如下:
python d_smiles2pv.py \
--checkpoint './Pretrain/checkpoint_SPMM.ckpt' \
--input_file './data/2_PV2SMILES/pubchem_1k_unseen.txt'生成过程输出如下:
seed: 543
Creating dataset
Creating model
LOADING PRETRAINED MODEL..
load checkpoint from ./Pretrain/checkpoint_SPMM.ckpt
_IncompatibleKeys(missing_keys=[], unexpected_keys=['temp'])
==================================================
SMILES-to-PV generation...
SMILES-to-PV generation done
mean of 53 properties' normalized RMSE: 0.12519080936908722
mean r^2 coefficient of determination: 0.9206444979846248
==================================================生成过程占用显存 3 GB,用时大约 2 分钟。输入的这 1000 个分子的 53 个属性的归一化 RMSE 的平均值约为 0.125,表明这些输入分子和训练数据集的属性差异较小。
3.4 基于预训练模型的下游任务
项目提供给了三个基于预训练模型,继续训练的三个不同类型的任务,分别对应回归、二分类和多分类回归任务和使用 BACE 、BBBP 和 Clintox 数据集继续训练。
3.4.1 BACE
使用 BACE 数据集重新训练,作为回归任务的示例。使用的训练集、验证集和测试集数据分别为 ./data/4_MoleculeNet 文件夹中的 BACEC_train.csv 、BACEC_valid.csv 和 BACEC_test.csv 。通过 d_regression.py 训练模型,但脚本中并没有保存模型的步骤,我们添加保存模型的代码。
def main(args, config):
device = torch.device(args.device)
print('DATASET:', args.name)
...
total_time = time.time() - start_time
total_time_str = str(datetime.timedelta(seconds=int(total_time)))
print('Training time {}'.format(total_time_str))
print('DATASET:', args.name, '\tTest set MSE of the checkpoint with best validation MSE:', best_test)
改为:
def main(args, config):
device = torch.device(args.device)
print('DATASET:', args.name)
...
total_time = time.time() - start_time
total_time_str = str(datetime.timedelta(seconds=int(total_time)))
# save the final model
print('Saving model...')
name = args.name
torch.save({'state_dict': model.state_dict()}, f'checkpoint_{name}.ckpt')
print('Training time {}'.format(total_time_str))
print('DATASET:', args.name, '\tTest set MSE of the checkpoint with best validation MSE:', best_test)继续训练的具体命令如下:
python d_regression.py \
--checkpoint './Pretrain/checkpoint_SPMM.ckpt' \
--name 'bace' 训练设置 50 个 epoch,整个训练过程大约 6 分钟,显存占用约 3GB,训练好的模型保存为 ./checkpoint_bace.ckpt 。
3.4.2 BBBP
使用 BBBP 数据集重新训练,作为二分类任务的示例。使用的训练集、验证集和测试集数据分别为 ./data/4_MoleculeNet 文件夹中的 BBBP_train.csv 、BBBP_valid.csv 和 BBBP_test.csv 。通过 d_classification.py 训练模型,但脚本中并没有保存模型的步骤,我们添加保存模型的代码。
def main(args, config):
device = torch.device(args.device)
print('DATASET:', args.name)
...
total_time = time.time() - start_time
total_time_str = str(datetime.timedelta(seconds=int(total_time)))
print('Training time {}'.format(total_time_str))
print('DATASET:', args.name, '\tTest set AUROC of the checkpoint with best validation AUROC:', best_test)
修改为:
def main(args, config):
device = torch.device(args.device)
print('DATASET:', args.name)
...
total_time = time.time() - start_time
total_time_str = str(datetime.timedelta(seconds=int(total_time)))
print('Training time {}'.format(total_time_str))
print('DATASET:', args.name, '\tTest set AUROC of the checkpoint with best validation AUROC:', best_test)
print('Saving model...')
name = args.name
torch.save({'state_dict': model.state_dict()}, f'checkpoint_{name}.ckpt')继续训练的具体命令如下:
python d_classification.py \
--checkpoint './Pretrain/checkpoint_SPMM.ckpt' \
--name 'bbbp'训练设置 50 个 epoch,整个训练过程大约 1 分钟,显存占用约 2 GB,训练好的模型保存为 ./checkpoint_bbbp.ckpt 。
3.4.3 Clintox
使用 Clintox 数据集重新训练,作为多分类任务的示例。使用的训练集、验证集和测试集数据分别为 ./data/4_MoleculeNet 文件夹中的 Clintox_train.csv 、Clintox_valid.csv 和 Clintox_test.csv 。通过 d_classification_multilabel.py 训练模型,但脚本中并没有保存模型的步骤,我们添加保存模型的代码。
def main(args, config):
device = torch.device(args.device)
print('DATASET:', args.name)
...
total_time = time.time() - start_time
total_time_str = str(datetime.timedelta(seconds=int(total_time)))
print('Training time {}'.format(total_time_str))
print('DATASET:', args.name, '\tTest set AUROC of the checkpoint with best validation AUROC:', best_test)改为:
def main(args, config):
device = torch.device(args.device)
print('DATASET:', args.name)
...
total_time = time.time() - start_time
total_time_str = str(datetime.timedelta(seconds=int(total_time)))
print('Training time {}'.format(total_time_str))
print('DATASET:', args.name, '\tTest set AUROC of the checkpoint with best validation AUROC:', best_test)
print('Saving model...')
name = args.name
torch.save({'state_dict': model.state_dict()}, f'checkpoint_{name}.ckpt')继续训练的具体命令如下:
python d_classification_multilabel.py \
--checkpoint './Pretrain/checkpoint_SPMM.ckpt' \
--name 'clintox'训练设置 50 个 epoch,整个训练过程大约 1 分钟,显存占用约 2 GB,训练好的模型保存为 ./checkpoint_clintox.ckpt 。
3.5 反应预测任务
反应预测任务包括正向和反向预测,使用的是 USPTO-480k 和 USPTO-50k 数据集。
3.5.1 正向反应预测(无束流搜索)
基于训练好的模型,batch_size 设置为 128 ,epoch 设置为 30。具体命令如下:
python d_rxn_prediction.py \
--checkpoint './Pretrain/checkpoint_SPMM.ckpt' \
--mode 'forward' \
--n_beam 1 训练过程中显存最大占用约 24 GB,训练用时约 20 个小时,训练好的模型保存在 ./output/RXN/checkpoint_best.pth,为了和反向(逆)反应进行区分,把文件重命名为 checkpoint_best_forward.pth 。
3.5.2 反向(逆)反应预测
基于训练好的模型,batch_size 设置为 128 ,epoch 设置为 30。具体命令如下:
python d_rxn_prediction.py \
--checkpoint './Pretrain/checkpoint_SPMM.ckpt' \
--mode 'retro' \
--n_beam 3训练过程中显存最大占用约 24 GB,训练用时约 18 个小时,训练好的模型保存在 ./output/RXN/checkpoint_best.pth,为了和正向反应进行区分,把文件重命名为 checkpoint_best_retro.pth 。
3.6 预训练模型训练
python SPMM_pretrain.py \
--data_path './data/1_Pretrain/pretrain_50m.txt'运行输出:
Initializing distributed: GLOBAL_RANK: 0, MEMBER: 1/1
----------------------------------------------------------------------------------------------------
distributed_backend=nccl
All distributed processes registered. Starting with 1 processes
----------------------------------------------------------------------------------------------------
You are using a CUDA device ('NVIDIA GeForce RTX 3090') that has Tensor Cores. To properly utilize them, you should set `torch.set_float32_matmul_precision('medium' | 'high')` which will trade-off precision for performance. For more details, read https://pytorch.org/docs/stable/generated/torch.set_float32_matmul_precision.html#torch.set_float32_matmul_precision
/home/wufeil/miniconda3/envs/SPMM/lib/python3.10/site-packages/pytorch_lightning/callbacks/model_checkpoint.py:615: UserWarning: Checkpoint directory ./Pretrain exists and is not empty.
rank_zero_warn(f"Checkpoint directory {dirpath} exists and is not empty.")
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
| Name | Type | Params
--------------------------------------------------------
0 | text_encoder | BertForMaskedLM | 100 M
1 | property_proj | Linear | 196 K
2 | text_proj | Linear | 196 K
3 | itm_head | Linear | 3.1 K
4 | property_embed | Linear | 1.5 K
5 | property_encoder | BertModel | 42.9 M
6 | property_mtr_head | Sequential | 592 K
7 | property_encoder_m | BertModel | 42.9 M
8 | property_proj_m | Linear | 196 K
9 | text_encoder_m | BertForMaskedLM | 100 M
10 | text_proj_m | Linear | 196 K
--------------------------------------------------------
144 M Trainable params
143 M Non-trainable params
288 M Total params
1,152.596 Total estimated model params size (MB)
Epoch 0: 0%| | 129/25000000 [00:43<2360:04:28,
四、总结
在本项工作中,作者提出了一种基于 Transformer 的多模态化学基础模型 SPMM。该模型能够进行分子结构和性质的双向生成/预测,并可用于诸如反应预测等单模态任务。SPMM 不仅在多模态挑战中表现出色,还为 SMILES 的单模态任务提供了有用的表示,可以进一步微调以适应许多分子下游任务。
尽管 SPMM 的表现显著,但仍有一些改进空间。其中之一是使用 SMILES 表示法。虽然 SMILES 可以包含分子二维结构的完整信息,但原子和键如何连接的信息仅隐含存在。而且,分子结构的细微变化会导致 SMILES 的巨大变化。分子表示的另一种广泛使用的形式是图格式,它包含明确的邻接矩阵信息,可以作为 SMILES 的替代品。
当前 SPMM 的另一个局限是使用的 53 个性质对于给定分子立体化学的变化是不变的。已知考虑立体化学在许多生化任务中起着关键作用。然而,由于这 53 个性质的值在不同的立体异构体中保持不变,它们无法提供任何立体化学信息的知识。这导致不同立体异构体的 SMILES 编码器输出趋于一致,因为对比损失将它们与相同的 PV 特征对齐。这是降低 SPMM 在 MoleculeNet 任务中表现的主要因素,可以通过使用更多反映分子立体化学的性质来解决。
我们那测试下来,SPMM 模型的结果与文章是相符的,代码没有错误,很好复现,值得进一步开发。


















 137
137

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











