







CMD-GEN 是Coarse-grained and Multi-dimensional Data-driven molecular generation的缩写。CMD-GEN 结合了蛋白质口袋信息和药效团特征的分子生成模型,发表期刊为 Commun Biol 8, 422 (2025)。

基于结构的药物设计旨在通过分析靶标结构来创造具有理想活性和性质的化合物。近年来,深度生成模型推动了针对特定结构的分子生成研究。然而,现有方法普遍受限于药物数据的不足,导致生成的分子性质欠佳、构象稳定性差。此外,这些方法往往忽视结合口袋的相互作用机制,在选择性抑制剂设计方面存在局限。为解决这些挑战,作者开发了一个名为"粗粒度多维数据驱动的分子生成"(CMD-GEN)的新框架。该框架通过扩散模型采样的粗粒度药效团点,将配体-蛋白质复合物与类药分子进行关联,有效扩充了训练数据。其分层架构将口袋内的三维分子生成分解为药效团点采样、化学结构生成和构象匹配三个步骤,显著提升了生成分子稳定性。基准测试表明 CMD-GEN 在分子性质优化方面优于现有方法,并实现了药物相似性的精准调控。在合成致死靶点案例研究中,CMD-GEN 展现出卓越性能。通过 PARP1/2 抑制剂的湿实验验证,进一步证实了该框架在设计选择性抑制剂方面的巨大潜力。
一、模型简介
当前,研究者尝试引入先验知识作为生成条件,包括分子片段、药效团特征、分子-靶标相互作用以及各类分子属性,通过引导模型在生成过程中逼近具有"生物活性"的分子。
但这类条件下生成的分子构象物理意义不明确,常与晶体构象存在偏差。此外,现有模型对特定设计需求(如双靶点抑制剂或高选择性抑制剂)的适配性仍有待提升。问题的核心在于药物数据固有的稀缺性与高噪声特性。将其他领域(如文本生成与图像识别)已验证有效的技术路径直接迁移至药物设计领域面临显著挑战。同时,现有模型多基于单一同质数据集训练,限制了先进算法的潜力。回顾 AlphaFold2 的成功经验,其突破源于共进化策略的引入与多维数据的整合。在创新药物设计领域,亟需将物理模型等科学概念与多维数据融入人工智能框架。
基于现有方法学的优势与局限,并受粗粒度分子动力学方法启发,作者提出了一种创新的基于结构的三维分子生成框架。通过将复杂问题解耦为子任务,将分子转化为药效团点云,以药效团为媒介,结合扩散模型与 transformer 编解码架构,建立有限的三维蛋白-配体复合物结构与海量药物分子序列间的关联映射。这种分层递进的策略实现了具有潜在生物活性分子的渐进式生成。同时,通过整合分子属性条件、分子对接与微调技术的迭代模型训练,实现了面向预设靶点的特异性活性分子生成。
CMD-GEN 依托药效团点云的桥梁作用,生成的分子实体可与靶标口袋无缝对接,得到具有明确物理意义的三维分子。此外,通过引入药效团点云的匹配分析,模型在选择性抑制剂或双靶点抑制剂生成等任务中展现出独特优势。通过PARP1/2 高选择性抑制剂的湿实验验证,证明 CMD-GEN 是一个强有力药物设计工具。该框架的提出丰富了现有药物研发工具箱,为分子生成与预测研究提供了新的视角。
二、模型结构与性能
2.1 模型结构
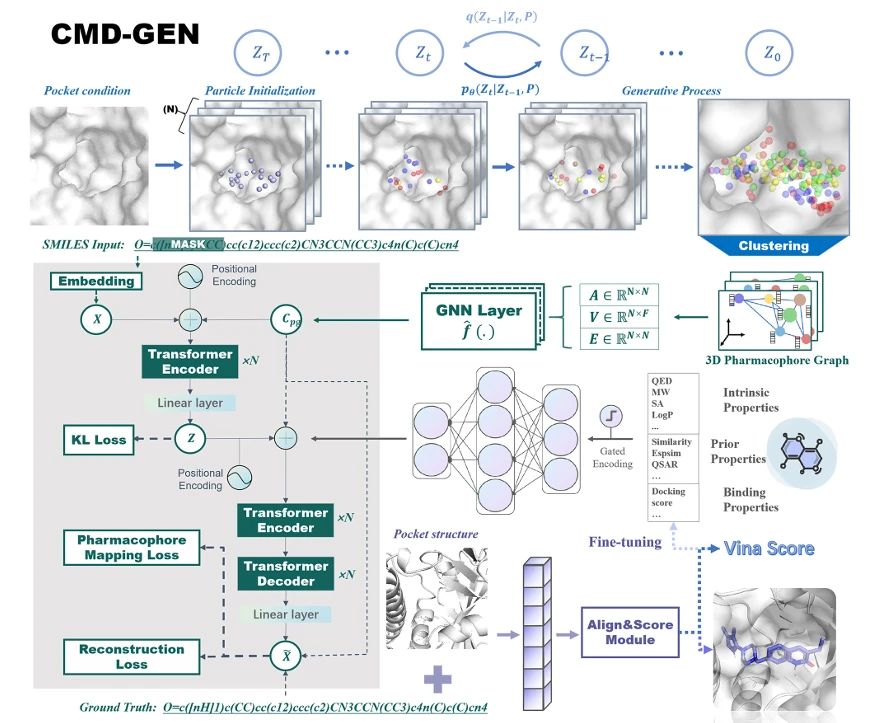
CMD-GEN 采用了分步架构,将口袋内的三维分子生成过程分解为以下几个紧密集成的主要模块:
(1)口袋条件三维药效团采样模块 (Pocket-conditioned three-dimensional pharmacophore sampling module): 该模块是 CMD-GEN 的第一步,其功能是在蛋白质口袋的约束下生成粗粒度的配体三维药效团点。该模块利用扩展的等变去噪扩散概率模型 (equivariant DenoisingDiffusion Probabilistic Model),通过模拟药效团的潜在三维坐标在口袋内的分布进行采样,蛋白质口袋的信息(3D 几何坐标和原子类型特征)在去噪过程的每一步都作为固定的三维上下文。设定的药效团类别包括芳香性、疏水性、可定位性、可负电性、受体、供体、聚集疏水基团和其他。此模块使用 crossdock 作为训练集。
(2)基于门控条件机制和药效团的分子生成模块 (Gating condition mechanism and pharmacophore-based molecular generation module - GCPG): 该模块的作用是将采样的重要药效团点云转化为化学结构,它基于 Transformer 编码器-解码器架构,利用药效团作为桥梁,学习药效团和分子之间复杂的映射关系,一个关键的创新点是门控条件机制的引入,该机制旨在优化或微调指定的分子性质,例如分子量 (MW)、LogP、QED 和 SA 等,从而提高分子生成过程的实用性和效率。此模块使用 ChEMB 作为训练集。
(3)基于药效团比对的分子结合构象生成模块 (Binding conformation generation module based on pharmacophore alignment): 在前两个阶段分别生成了药效团点云和化学结构后,该模块通过简单的叠加策略来实现预测的分子结合构象。为了解决模型随机性可能导致药效团无法完美对齐的问题,该模块引入了容差参数,允许一定数量的药效团点不匹配,从而可以生成多个计算上可行的配体结合构象。
根据作者的模型结构示意图,CMD-GEN 可以完成分子骨架跃迁、片段替换任务。
2.2 模型性能
在药效团的采样方面,CMD-GEN 采样的药效团与训练集的分布非常相似,如下图:
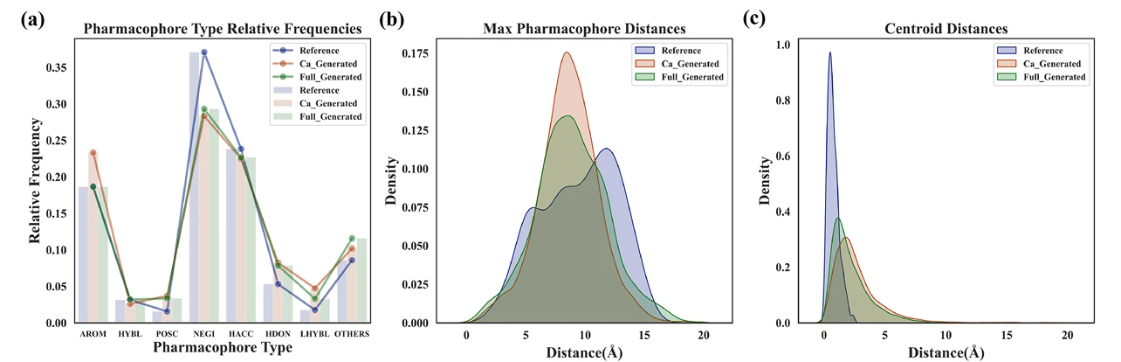
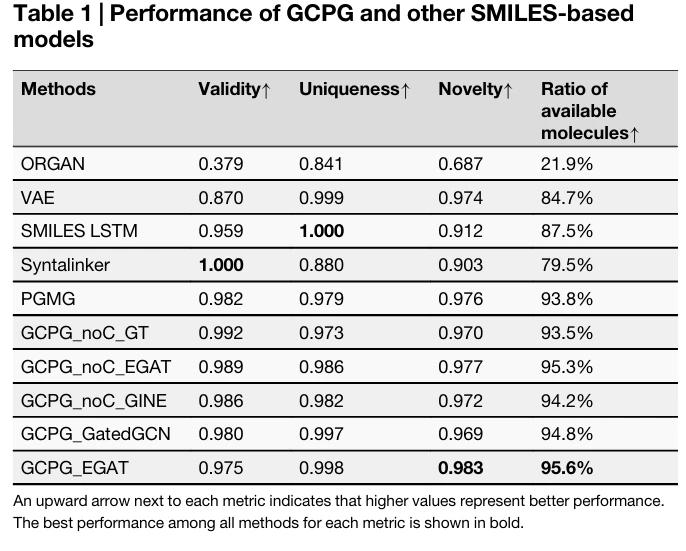
在药效团条件和物理性质门控下,CMD-GEN 展现出较优秀的有效率、新颖率和可得率。
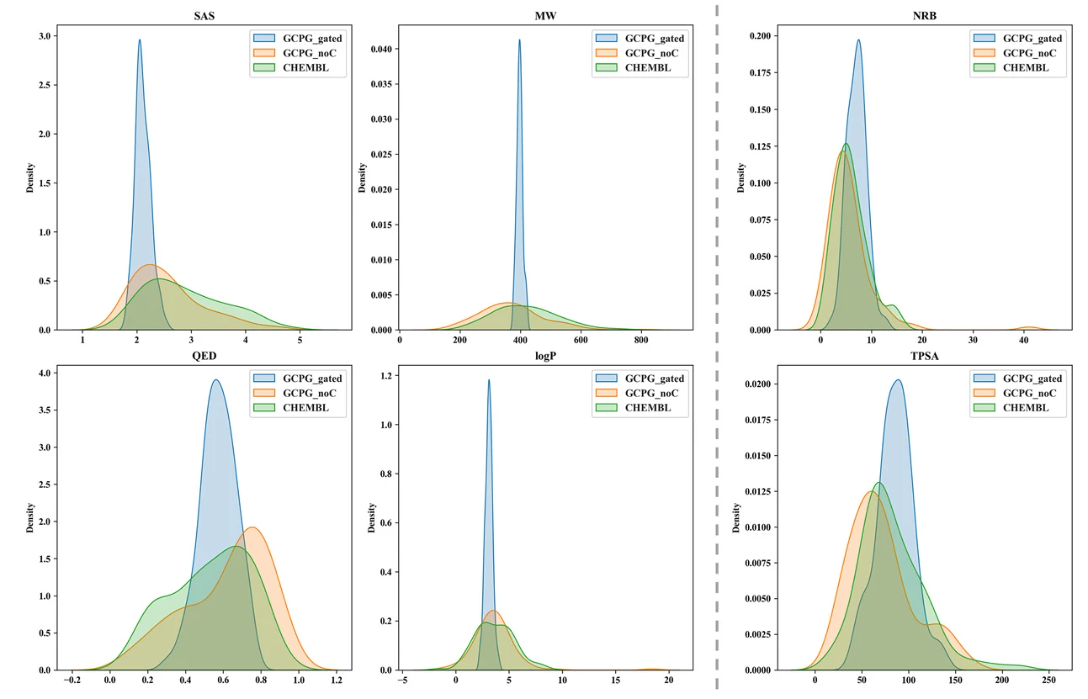
生成的分子的物理化学性质分布明显小于训练集,能满足特定物理化学性质的分子生成任务。
在实际靶点 PARP1 的分子生成任务上,CMD-GEN 生成的分子与已知活性分子具有类似的结合模式和结构:
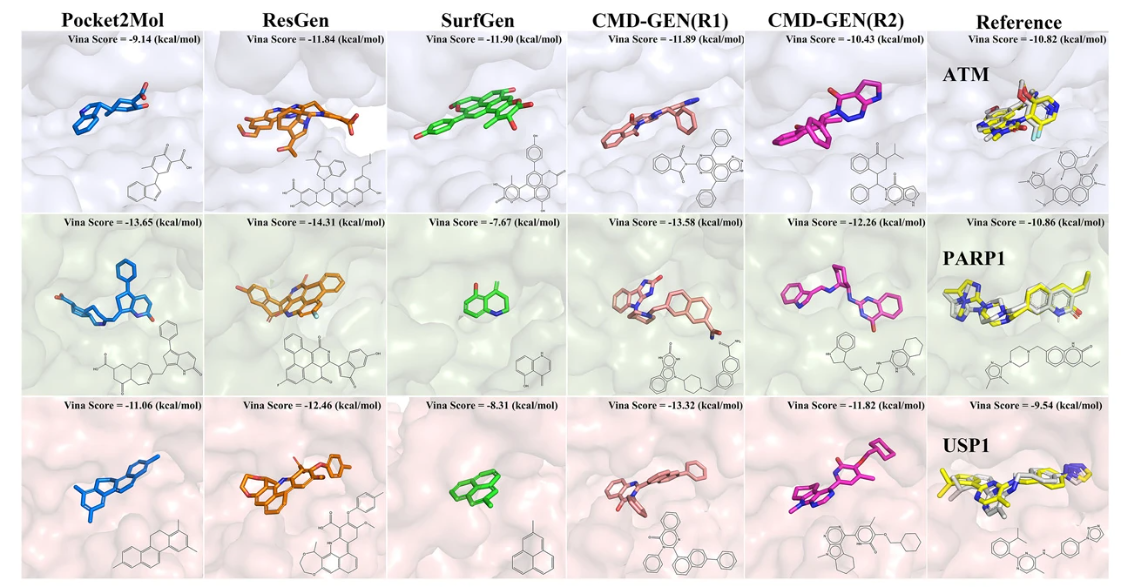
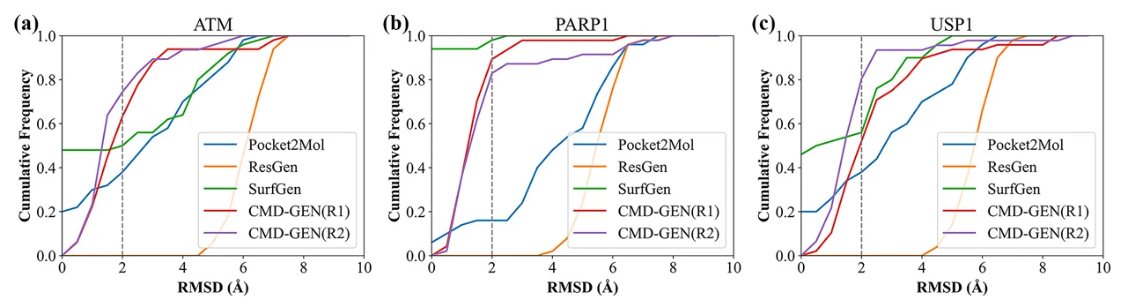
在ATM, PARP1, USP1靶点上,对生成的分子进行重对接,能较好保持原有的结合模式。
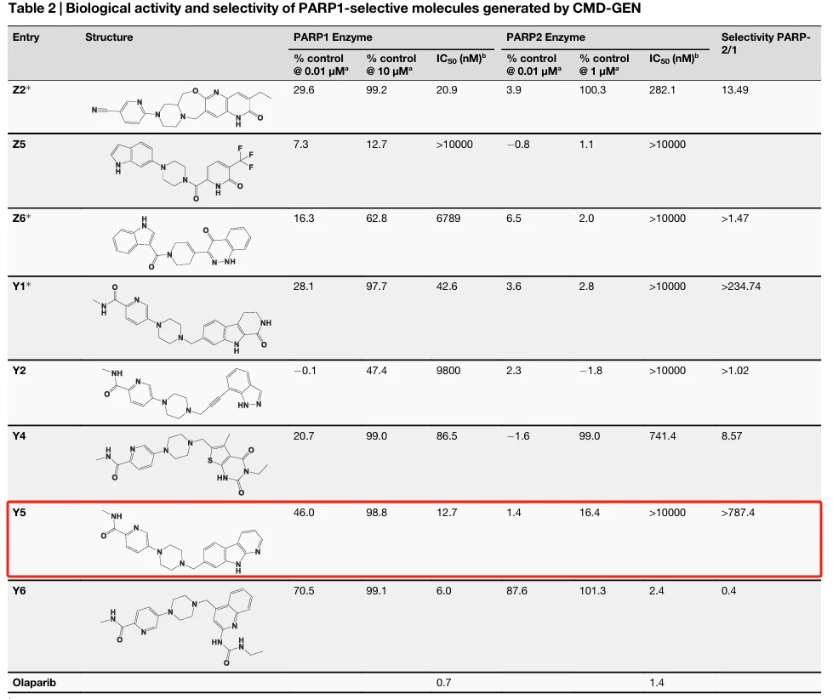
基于药效团分析,进行的分子生成,经过虚拟筛选和人工筛选后,实际测试的10个分子中,存在一个显著选择性,且 IC50=12.7nM 的新骨架分子。
三、CMD-GEN 评测
粗粒化多维数据驱动分子生成(CMD-GEN)通过利用扩散模型采样的粗粒度药效团点,将三维配体-蛋白质复合物数据与二维类药物分子数据相桥接,从而丰富生成模型的训练数据。通过分层式架构,将口袋内三维分子生成解耦为粗粒度药效团点采样、化学结构生成和构象对齐三个阶段,避免了基于深度生成模型直接生成分子构象所固有的不稳定性问题。
注:当前开源的 CMD-GEN 项目,是初始工作版本,并不是完整版。
3.1 安装环境
根据项目提供的 yaml 文件安装项目运行环境,通过下面命令创建 DiffPhar 和 GCPG 环境:
conda env create -f ./DiffPhar/env/environment_diffphar.ymlpip 在安装 torch 的时候没有找到指定版本的库,报错如下:
Pip subprocess error:
ERROR: Could not find a version that satisfies the requirement torch==1.12.1+cu116 (from versions: 1.11.0, 1.12.0, 1.12.1, 1.13.0, 1.13.1, 2.0.0, 2.0.1, 2.1.0, 2.1.1, 2.1.2, 2.2.0, 2.2.1, 2.2.2, 2.3.0, 2.3.1, 2.4.0, 2.4.1, 2.5.0, 2.5.1, 2.6.0, 2.7.0)
ERROR: No matching distribution found for torch==1.12.1+cu116
failed
CondaEnvException: Pip failed
激活环境,手动安装指定版本的 Pytorch 相关的库,命令如下:
conda activate DiffPhar
pip install torch==1.13.1+cu116 torchvision==0.14.1+cu116 torchaudio==0.13.1 --extra-index-url https://download.pytorch.org/whl/cu116
pip install torch-scatter -f https://data.pyg.org/whl/torch-1.13.1+cu116.html
通过下面命令创建 GCPG 环境:
conda env create -f ./GCPG/env/environment_gcpg.yml
通过 pip 没有找到指定版本的 dgl-cu116==0.9.0 ,报错如下:
Pip subprocess error:
ERROR: Could not find a version that satisfies the requirement dgl-cu116==0.9.0 (from versions: none)
ERROR: No matching distribution found for dgl-cu116==0.9.0
failed
CondaEnvException: Pip failed 进入 GCPG 环境,手动下载 dgl-cu116==0.9.0 ,命令如下:
conda activate GCPG
pip install dgl-cu116==0.9.0 -f https://data.dgl.ai/wheels/repo.html手动安装其他库:
pip install rdkit-pypi==2022.9.5 requests==2.31.0 scipy==1.10.1 urllib3==2.0.4
pip install torch==1.12.0+cu116 torchvision==0.13.0+cu116 torchaudio==0.12.0 --extra-index-url https://download.pytorch.org/whl/cu116
至此, DiffPhar 和 GCPG 两个运行环境已经安装好了。这里测试安装了项目运行的两个环境,但在生成部分我们把所有的库都安装在 GCPG ,只在这一个环境中测试项目。
下载模型权重
项目提供测试代码和训练好的权重文件,保存在 Zenodo,链接为:https://zenodo.org/records/13841142 ,网页截图如下:
下载后的 CMD-GEN.zip 文件上传到项目目录后解压:
unzip CMD-GEN.zip
项目提供的 checkpoints 保存在 ./CMD-GEN/code/CMD-GEN/DiffPhar/checkpoints 文件夹中,目录如下。文件夹中分别包含 ca_atom 和 full_atom 两个模型。
.
|-- ca_atom
| `-- best-model-epoch=epoch=472.ckpt
`-- full_atom
`-- best-model-epoch=epoch=281.ckpt
2 directories, 2 files3.2 基于结合口袋的三维药效团采样模块
为方便使用,这里测试的内置案例和自定义案例统一在 GCPG 环境中运行。
注:在运行过程中,出现了较多的错误。有些在文中已经指出,还有一些错误,没有写下来。如果有需要请下载附件《CMD-GEN 的可运行代码+完整测评文档》,里面记录了详细了测评过程。
3.2.1 内置案例
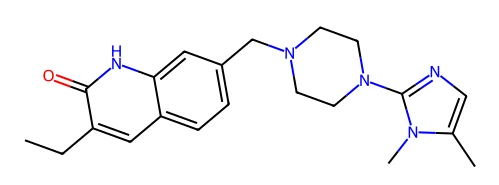
项目举例的内置案例是 7ONS.pdb 蛋白质,手动从 PDB 数据库中下载蛋白结构,该结构是PARP1催化结构域与基于异喹啉酮的抑制剂形成的复合物。使用 pymol 删去溶剂,只保留下 A 链结构。配体在口袋中的位置如下:
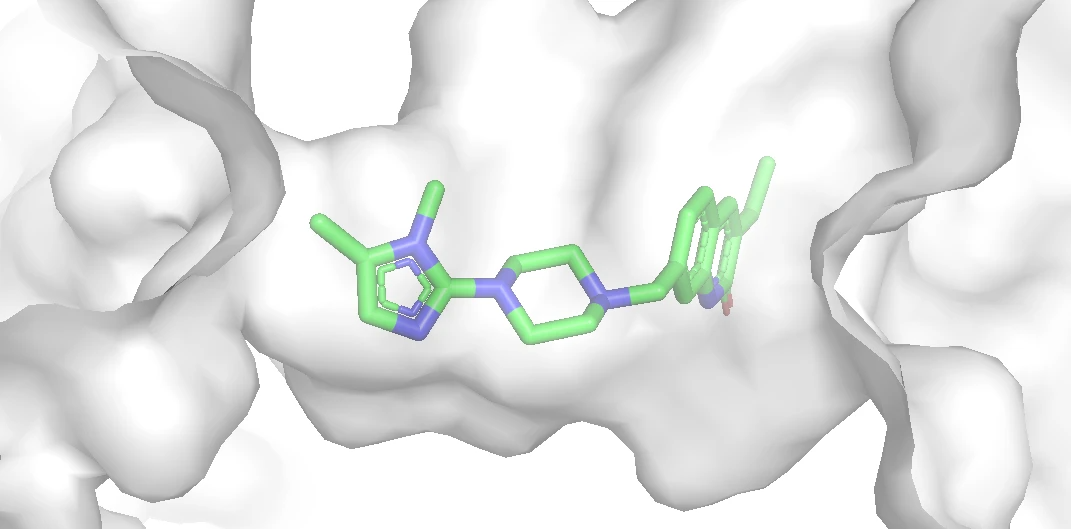
配体的 2D 结构如下:
3.2.1.1 生成药效团点
在获得训练好的权重后,可以使用 generate_phars.py 脚本生成药效团点。该脚本需要提供以下参数:预训练检查点文件路径、目标蛋白的PDB文件路径以及参考配体(格式为链:索引)。命令:
python ./DiffPhar/generate_phars.py ./CMD-GEN/code/CMD-GEN/DiffPhar/checkpoints/ca_atom/best-model-epoch=epoch=472.ckpt \
--num_nodes_phar 10 \
--pdbfile ./data/7ONS_A.pdb \
--ref_ligand A:1101 \
--outdir ./generate_phars其中,
- checkpoint 用来指定使用的 checkpoint 文件;
- --num_nodes_phar 设置药效团采样的个数;
- --pdbfile 传入蛋白和配体的复合物结构;
- --ref_ligand 指定配体的位置,比如:A 链的 1101 残基。
注:运行命令有安装包错误,直接安装完成就好。
生成 ./phar_to_coords_no_tensor_PI3K_dul.json 文件,该文件记录了药效团特征到空间坐标的映射对应关系,但没有使用张量的数据格式,而使用的是字典这种基本数据结构。运行采用个数设置为 10,json 文件中包含Molecule_1 到 Molecule_10 的信息,部分内容如下所示:
{"Molecule_1": {"Acceptor": [[15.544816970825195, 45.20138931274414, 4.3889007568359375], [8.914205551147461, 43.050933837890625, 7.957984924316406], [15.338077545166016, 45.143951416015625, 4.150893211364746], [9.139785766601562, 44.87507247924805, 6.774946212768555], [15.439044952392578, 45.459800720214844, 7.393172740936279], [11.756556510925293, 38.702598571777344, 6.924417018890381], [13.99411678314209, 41.351890563964844, 8.806380271911621], [13.049095153808594, 42.59394836425781, 7.04973840713501], [10.397403717041016, 46.24335479736328, 8.813750267028809]], "PosIonizable": [[14.359405517578125, 43.5037956237793, 6.7066545486450195], [12.286407470703125, 42.35445785522461, 5.878037452697754]], "Donor": [[7.17282247543335, 43.94087600708008, 6.4164252281188965], [11.45179557800293, 44.05781555175781, 7.052504062652588], [10.897769927978516, 45.284698486328125, 7.731184959411621], [13.257363319396973, 43.88445281982422, 5.581877708435059]], "LumpedHydrophobe": [[15.93664264678955, 42.71268844604492, 7.182422161102295], [7.072929859161377, 41.463260650634766, 7.5018134117126465], [13.661638259887695, 42.392967224121094, 7.879695892333984]], "Aromatic": [[9.787681579589844, 41.68983459472656, 8.030027389526367], [15.304008483886719, 42.48984909057617, 7.353493690490723]]},
...}处理的药效团类型如下表:
| 药效团名称 | 含义 | 描述 |
| Acceptor | 氢键受体 | 能接受氢键的原子或基团(如羰基氧、醚氧) |
| PosIonizable | 可电离正电荷 | 可质子化带正电的基团(如氨基 -NH₃⁺) |
| Donor | 氢键供体 | 能提供氢键的原子或基团(如羟基 -OH、氨基 -NH₂) |
| LumpedHydrophobe | 疏水基团 | 非极性疏水区域(如烷基链、芳香环疏水面) |
| Aromatic | 芳香环 | 平面共轭芳香体系(如苯环、吡啶环) |
3.2.1.2 进行降维和聚类
为识别关键药效团点,将采用高斯混合模型(GMM)进行降维处理。需要修改 ./DiffPhar/get_phar/GMM_json.py 中的参数,打开 ./DiffPhar/get_phar/GMM_json.py 脚本,把第 12 行的代码从
input_file_path = 'phar_to_coords_no_tensor_PARP1.json' 修改为:
input_file_path = '/workspace/GuanXL/projects/CMD_GEN/phar_to_coords_no_tensor_PI3K_dul.json'
第 26 行代码设置聚多少个类,默认为 7 ,可以尝试 5、6 等。这个需要根据实际情况进行微调。
# Choose the number of clusters (you may need to tune this based on your data)
n_clusters = 7 # You can try values like 5, 6, or 7
运行该脚本,命令如下:
python ./DiffPhar/get_phar/GMM_json.py运行输出为 output.posp 文件,文件内容如下。该文件分别提取出聚类的 7 个簇概率最大的药效团种类和簇的中心坐标。比如 HDON 13.62 43.23 6.47 表示第一个簇中概率最大的药效团是氢键供体(HDON),簇的中心坐标为 13.62 43.23 6.47 。该文件用于提供 3D 分子生成模型的指导信息:
HDON 13.62 43.23 6.47
LHYBL 8.23 44.60 6.97
HDON 11.46 43.26 7.40
AROM 15.50 43.46 6.77
LHYBL 11.74 41.20 6.45
POSC 8.70 40.59 6.75
HACC 15.17 45.24 4.63根据处理的药效团文件,理想的生成分子应该包含:
(1)两个氢键供体基团(匹配HDON位点)
(2)一个氢键受体基团(匹配HACC)
(3)带正电基团(如叔胺,匹配POSC)
(4)疏水片段(匹配LHYBL区域)
(5)芳香环系统(匹配AROM)
药效团的空间排布要求如下:
疏水区(LHYBL)───氢键供体(HDON)───芳香区(AROM)
| | |
正电基团(POSC) 氢键供体(HDON) 氢键受体(HACC-深层)
| |
疏水区(LHYBL)───────┘
3.2.1.3 生成分子 SMILES
通过门控条件机制与基于药效团的分子生成模块(GCPG)可以生成药效团与性质约束的分子,当前支持的性质如下:
MW, logP, QED, SAS, RotaNumBonds当前默认支持的药效团类型包括:
(1)AROM:芳香环
(2)POSC:阳离子基团
(3)HACC:氢键受体
(4)HDON:氢键供体
(5)HYBL:疏水基团(环状)
(6)LHYBL:疏水基团(非环状)
在.posp文件中的三维坐标将首先用于计算每两个点之间的欧几里得距离,然后这些距离将被映射为基于最短路径的距离。修改 ./GCPG/generate.py 中的参数,以便循环生成期望的化学结构。参数设置如下:
MW_min, MW_max, MW_step = 400, 400, 1
logP_min, logP_max, logP_step = 4, 4, 1
QED_min, QED_max, QED_step = 0.6, 0.6, 1
SAS_min, SAS_max, SAS_step = 4, 4, 1
RotaNumBonds_min, RotaNumBonds_max, RotaNumBonds_step = 4, 4, 1设置好参数之后,运行 ./GCPG/generate.py 脚本来生成分子。该脚本的运行参数如下所示:
python generate.py \
[-h] \
[--n_mol N_MOL] \
[--device DEVICE] \
[--filter] \
[--batch_size BATCH_SIZE] \
[--seed SEED] \
input_path \
output_dir \
model_path \
tokenizer_path其中,
[-h] 显示帮助信息;
[--n_mol N_MOL] 用来指定每个药效团文件生成分子的数目,默认为 200;
[--device DEVICE] 指定模型运行的设备,可选 cpu 或者 cuda,默认为 cpu ;
[--filter] 设置是否只保存唯一有效的分子;
[--batch_size BATCH_SIZE] 设置模型运行的批次大小,默认为 128 ;
[--seed SEED] 设置随机种子,默认为 1;
input_path 指定输入文件路径,如果指定文件夹,其中所有后缀为 .edgep 或 .posp 的文件都会被处理;
output_dir 指定输出文件的保存路径;
model_path 指定模型文件,为 .pth 文件;
tokenizer_path 指定保存 .pkl 格式的分词器文件的路径。
运行命令如下:
python ./GCPG/generate.py \
./output.posp \
./gen_result/ \
./CMD-GEN/code/CMD-GEN/GCPG/checkpoints/fold0_epoch32.pth \
./tokenizer_r_iso.pkl \
--filter \
--device cpu由于项目没有提供 tokenizer_r_iso.pkl 文件,需要根据训练集、验证集、测试集的数据生成该文件。
项目提供预处理好的数据集在 ./CMD-GEN/dataset/source_data/gcpg/data_prepare.zip,解压该压缩包:
unzip ./CMD-GEN/dataset/source_data/gcpg/data_prepare.zip数据解压到 ./CMD-GEN/dataset/source_data/gcpg/data_prepare 文件夹中,内容如下:
|-- chembl24_canon_test.pickle
|-- chembl24_canon_train.pickle
|-- chembl24_canon_valid.pickle
|-- chembl24_test_properties.pickle
|-- chembl24_train_properties.pickle
|-- chembl24_valid_properties.pickle
|-- chembl33_filtered_test_data.pickle
|-- chembl33_filtered_train_data.pickle
|-- chembl33_filtered_valid_data.pickle
|-- data_cond.py
|-- docking_atm_epoch1_properties.pickle
|-- docking_parp_epoch1_properties.pickle
|-- docking_usp_epoch1_properties.pickle
|-- filter_data.py
|-- fpscores.pkl.gz
`-- smiles2ppgraph.py
0 directories, 16 files其中,
(1)chembl33_filtered_train_data.pickle
(2)chembl33_filtered_valid_data.pickle
(3)chembl33_filtered_test_data.pickle
分别为处理好的训练集、验证集、测试集。
运行命令:
python ./GCPG/generate.py \
./output.posp \
./gen_result/ \
./CMD-GEN/code/CMD-GEN/GCPG/checkpoints/fold0_epoch32.pth \
./tokenizer_r_iso.pkl \
--filter \
--device cpu
注:V1 版本的模型文件是正常的,V2 版本的模型文件是破损文件,使用 V1 版本的模型来生成模型。
命令正常运行,几秒钟就完成了分子生成。输出文件保存在 ./gen_result 文件夹中,生成分子以 SMILES 的形式保存在 ./gen_result/output_result.txt 文件中是,内容如下所示:
CC(NC1CCC(c2nc3ccnn3c(=O)[nH]2)c2ccccc21)c1ccccc1
CCn1cc(Nc2cccc3[nH]c(=O)cc(N4CCC(C)c5ccccc54)c23)nn1
CCCN1CC2CCCCC2CC1n1c(=O)n(-c2ccc[nH]2)c(=O)c2ccccc21
...
CCOC(=O)C1CC(c2ccc(F)cc2)NCC1c1cc(=O)c2ccccc2[nH]1
CCCc1[nH]n(-c2cc(=O)[nH]c3ccccc23)c(=O)c1C1CC(C)CC(N)C1一共生成了 167 个分子 SMILES,生成命令传入了 --filter 参数,所以保存的分子都是唯一有效的分子。
3.2.1.4 基于药效团匹配的分子结合构象生成
可以通过运行 ./GCPG/align_dir/align_ligandpharm_modify.py 脚本基于药效团匹配,把筛选出来的分子 SMILES 转换成合适的分子构象。脚本传入的主要参数如下所示:
python ./GCPG/align_dir/align_ligandpharm_modify.py \
[-smi] \
[--smiles_path PATH] \
[--Input_path .psop_file] \
[--output_fir Path] \
[--phar_tolerance]其中,
-smi 传入药效团匹配的输入分子,一般为给定的参考配体的 SMILES ;
--smiles_path 传入前面生成的分子 SMILES,比如:./gen_result/output_result.txt ;
--Input_path 传入保存提供药效团空间排列数据的文件,比如:./output.posp
--output_fir 指定生成的分子构象保存的路径;
--phar_tolerance 表示药效团匹配的容忍度,即生成分子药效团特征和参考分子之间的最大允许偏差。1Å 以内一般认为严格匹配,1.0-1.5 Å 为中等匹配,1.5-2.5 Å 为宽松匹配。
注:说明文件中这里使用的脚本名称错误,写成了 generate.py,实际应该为 align_ligandpharm_modify.py 。
创建 ./gen_mol 文件夹,用来保存生成的分子构象。匹配药效团命令如下,后台挂起运行命令,打印内容记录在 align_ligandpharm_modify.log 文件中,并记录命令运行时间。
nohup bash -c "time python ./GCPG/align_dir/align_ligandpharm_modify.py \
--smiles_path ./gen_result/output_result.txt \
--input_path ./output.posp \
--output_dir ./gen_mol \
--phar_tolerance 1" > align_ligandpharm_modify.log 2>&1 &命令运行费时约 5 分钟,生成结果保存在 ./gen_mol 文件夹,目录如下所示:
.
|-- lig0_phar0
| `-- aligned_molecule_1.sdf
|-- lig101_phar0
| `-- aligned_molecule_1.sdf
...
|-- lig96_phar0
| `-- aligned_molecule_1.sdf
|-- lig97_phar0
| `-- aligned_molecule_1.sdf
`-- lig99_phar0
`-- aligned_molecule_1.sdf
125 directories, 125 files生成设置采样 200 个分子,经过去除重复分子、无效分子、药效团差异较大的分子,最终保存了 125 个分子构象,占比为 62.5% 。
3.2.2 自定义案例(3WZE)
我们把 3wze.pdb 蛋白作为自定义测试案例,配体在口袋中的构象如下:
口袋中分子的 2D 结构,如下:
3.2.2.1 生成药效团点
生成分子前,需要对给定配体进行药效团采样。从 PDB 库中下载的 3WZE.pdb 结构,使用 PyMol 删除溶剂,只保留蛋白和配体结构。创建 ./3WZE 文件夹,把预处理好的 3WZE.pdb 上传到该文件夹。
获取口袋中原始配体的药效团信息,配体在 A 链的 1201 位。手动修改脚本中输出文件名,把:
output_file_path = Path('phar_to_coords_no_tensor_PI3K_dul.json')修改为:
output_file_path = Path('phar_to_coords_no_tensor_3WZE.json')在 GCPG 中测试自定义案例,激活命令如下:
conda activate GCPG提取药效团信息的命令如下:
python ./DiffPhar/generate_phars.py ./CMD-GEN/code/CMD-GEN/DiffPhar/checkpoints/ca_atom/best-model-epoch=epoch=472.ckpt \
--num_nodes_phar 10 \
--pdbfile ./3WZE/3WZE.pdb \
--ref_ligand A:1201 生成 ./phar_to_coords_no_tensor_3WZE.json 文件,该文件记录了药效团特征到空间坐标的映射对应关系,但没有使用张量的数据格式,而使用的是字典这种基本数据结构。运行采用个数设置为 10,json 文件中包含Molecule_1 到 Molecule_10 的信息,部分内容如下。其中的药效团信息解释可以参考《3.2.1.1 生成药效团》这一小节。
{"Molecule_1": {"Donor": [[26.897369384765625, 22.588027954101562, 36.58668899536133], [25.424394607543945, 23.411911010742188, 36.32173156738281], [21.651912689208984, 28.379562377929688, 39.45027160644531], [27.229026794433594, 20.896453857421875, 35.111629486083984], [22.242109298706055, 23.401535034179688, 36.142974853515625], [27.070514678955078, 20.504852294921875, 34.43379592895508], [26.613433837890625, 20.604677200317383, 35.59125900268555]], "Hydrophobe": [[26.462291717529297, 22.462848663330078, 37.98717498779297], [23.948583602905273, 23.74520492553711, 37.52337646484375], [26.598440170288086, 23.24661636352539, 34.29133987426758], [25.00379180908203, 22.556135177612305, 36.74687957763672], [25.798084259033203, 22.796417236328125, 36.48051834106445], [23.29768943786621, 23.778961181640625, 38.92402648925781]], "Aromatic": [[22.66819190979004, 24.826061248779297, 38.30207061767578], [18.543304443359375, 26.563310623168945, 40.06367874145508]], "Acceptor": [[26.761276245117188, 20.624319076538086, 36.98097229003906], [24.110485076904297, 23.724000930786133, 36.752838134765625], [27.81268882751465, 20.71982192993164, 35.278602600097656]], "LumpedHydrophobe": [[20.212984085083008, 25.36630630493164, 38.920684814453125], [22.999309539794922, 24.191940307617188, 39.04466247558594]]},
...}3.2.2.2 进行降维和聚类
为识别关键药效团点,将采用高斯混合模型(GMM)进行降维处理。需要修改 ./DiffPhar/get_phar/GMM_json.py 中的参数,打开 ./DiffPhar/get_phar/GMM_json.py 脚本,把第 12 行的代码设置生成的 json 文件路径:
input_file_path = '/workspace/GuanXL/projects/CMD_GEN/phar_to_coords_no_tensor_3WZE.json' 修改脚本中的保存的 .posp 文件的路径,把:
# Writing to the .posp file
with open("output.posp", "w") as file:
for line in output_data:
file.write(line + "\n")修改为:
# Writing to the .posp file
with open("./3WZE/3WZE.posp", "w") as file:
for line in output_data:
file.write(line + "\n")运行该脚本,命令如下:
python ./DiffPhar/get_phar/GMM_json.py运行输出为 ./3WZE/3WZE.posp 文件,文件内容如下。该文件分别提取出聚类的 7 个簇概率最大的药效团种类和簇的中心坐标。比如 POSC 26.47 21.81 37.08 表示第一个簇中概率最大的药效团是带正电基团(POSC),簇的中心坐标为 26.47 21.81 37.08 。该文件用于提供 3D 分子生成模型的指导信息
POSC 26.47 21.81 37.08
AROM 19.93 25.78 38.81
POSC 25.91 21.94 35.43
HACC 21.71 24.93 37.71
HACC 20.24 28.04 39.73
HYBL 22.97 24.39 38.45
UNKNOWN 24.87 23.34 36.64根据处理的药效团文件,理想的生成分子应该包含:
(1)两个氢键受体基团(匹配 HACC )
(2)两个带正电基团(如叔胺,匹配 POSC )
(3)芳香环系统(匹配 AROM )
(4)疏水片段(匹配 HYBL 区域)
3.2.2.3 生成分子 SMILES
通过门控条件机制与基于药效团的分子生成模块(GCPG)可以生成药效团与性质约束的分子。在 .posp 文件中的三维坐标将首先用于计算每两个点之间的欧几里得距离,然后这些距离将被映射为基于最短路径的距离。修改 ./GCPG/generate.py 中的参数,以便循环生成期望的化学结构。参数设置如下:
MW_min, MW_max, MW_step = 400, 400, 1
logP_min, logP_max, logP_step = 4, 4, 1
QED_min, QED_max, QED_step = 0.6, 0.6, 1
SAS_min, SAS_max, SAS_step = 4, 4, 1
RotaNumBonds_min, RotaNumBonds_max, RotaNumBonds_step = 4, 4, 1运行命令如下:
python ./GCPG/generate.py \
./3WZE/3WZE.posp \
./3WZE/ \
./CMD-GEN/code/CMD-GEN/GCPG/checkpoints/fold0_epoch32.pth \
./tokenizer_r_iso.pkl \
--filter \
--device cpu命令正常运行,几秒钟就完成了分子生成。输出文件保存在 ./3WZE 文件夹中,生成分子以 SMILES 的形式保存在 ./3WZE/3WZE_result.txt 文件中是,内容如下所示:
CCN1C[N+]2(CCc3cc4cc(F)ccc4nc3C3CCCCC3)CCN1CC2
COc1ccc2sc(NC(S)=NC(C)C3N=CN3C3CCCCC3)nc2c1
COc1ccc2[nH]c(C3CC(C)CN3C([S-])=NC(C)c3ccccc3)nc2c1
...
COc1ccc2oc(C3NC(Nc4ccccc4Cl)=NC(C)=C3C#N)nc2c1
CC(NCC1CCCCN1)c1cc2c(nn1)-c1nc3ccc(F)cc3cc1CC2一共生成了 195 个分子 SMILES,生成命令传入了 --filter 参数,所以保存的分子都是唯一有效的分子。
3.2.2.4 基于药效团匹配的分子结合构象生成
可以通过运行 ./GCPG/align_dir/align_ligandpharm_modify.py 脚本基于药效团匹配,把筛选出来的分子 SMILES 转换成合适的分子构象。
创建 ./3WZE/gen_mol 文件夹,用来保存生成的分子构象。匹配药效团命令如下,后台挂起运行命令,打印内容记录在 align_ligandpharm_modify_3WZE.log 文件中,并记录命令运行时间。
nohup bash -c "time python ./GCPG/align_dir/align_ligandpharm_modify.py \
--smiles_path ./3WZE/3WZE_result.txt \
--input_path ./3WZE/3WZE.posp \
--output_dir ./3WZE/gen_mol \
--phar_tolerance 1" > align_ligandpharm_modify_3WZE.log 2>&1 &命令运行费时约 5 分钟,生成结果保存在 ./3WZE/gen_mol 文件夹,目录如下所示:
.
|-- lig0_phar0
| `-- aligned_molecule_1.sdf
|-- lig100_phar0
| `-- aligned_molecule_1.sdf
...
|-- lig97_phar0
| `-- aligned_molecule_1.sdf
|-- lig98_phar0
| `-- aligned_molecule_1.sdf
`-- lig9_phar0
`-- aligned_molecule_1.sdf
120 directories, 120 files
生成设置采样 200 个分子,经过去除重复分子、无效分子、药效团差异较大的分子,最终保存了 120 个分子构象,占比为 60% 。
3.2.2.5 评估生成分子
把每个生成分子的最佳对接打分作为分子属性写入到 SDF 文件中,所有生成分子按照打分升序排列(亲和力打分越好越靠前),合并到 sdf 中。
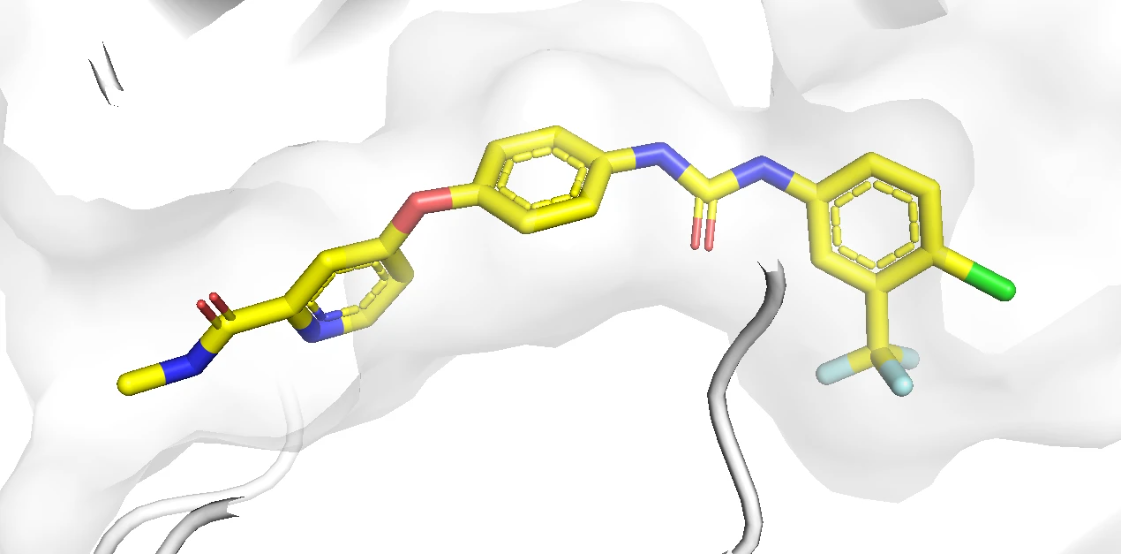
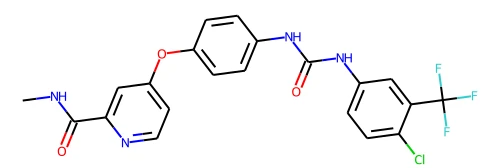
对接打分最好的 Top 3 分子的 pose 如下图(蓝色为口袋中的原始配体,紫红色是生成分子)。可以看出,这几个生成分子的原始构象都和蛋白口袋存在空间冲突。
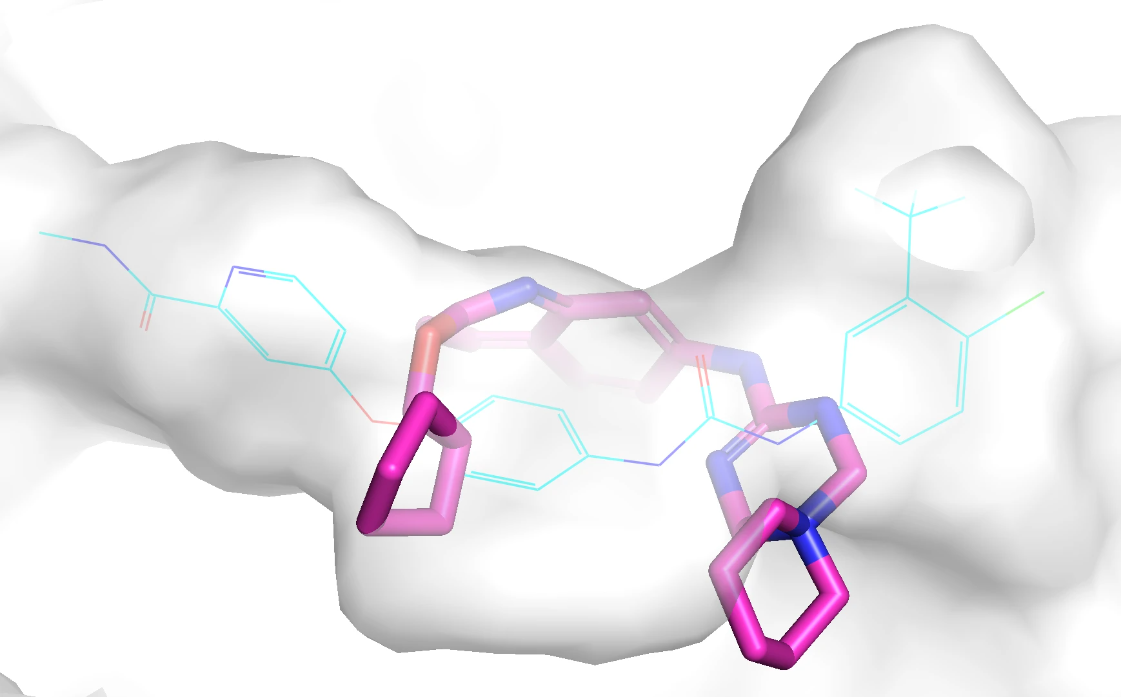
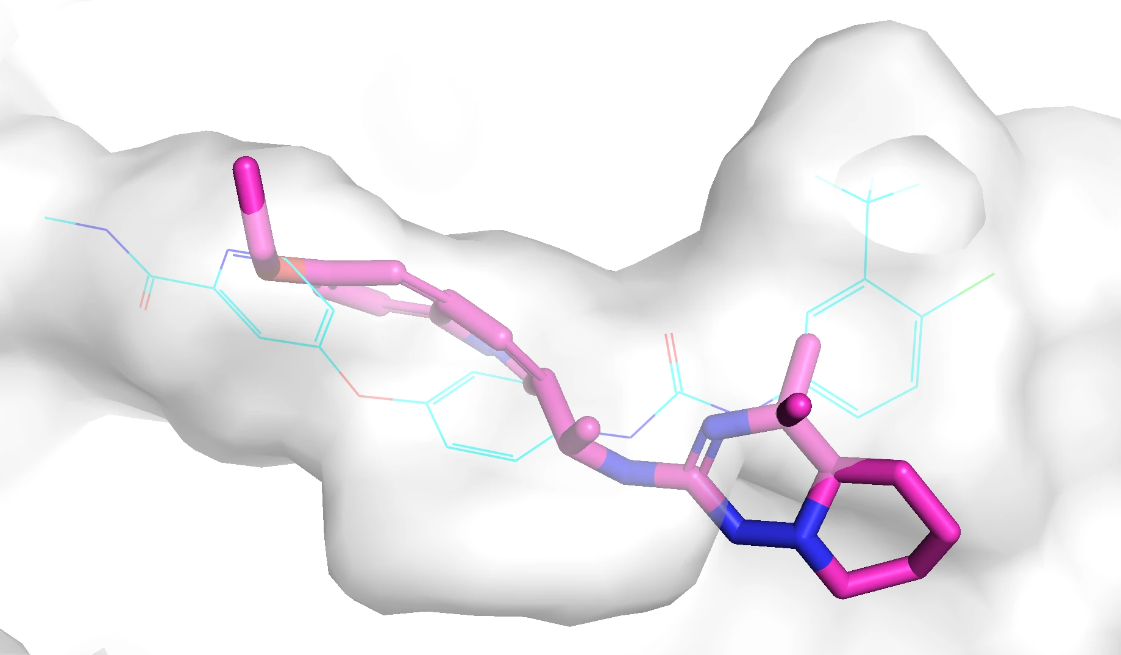
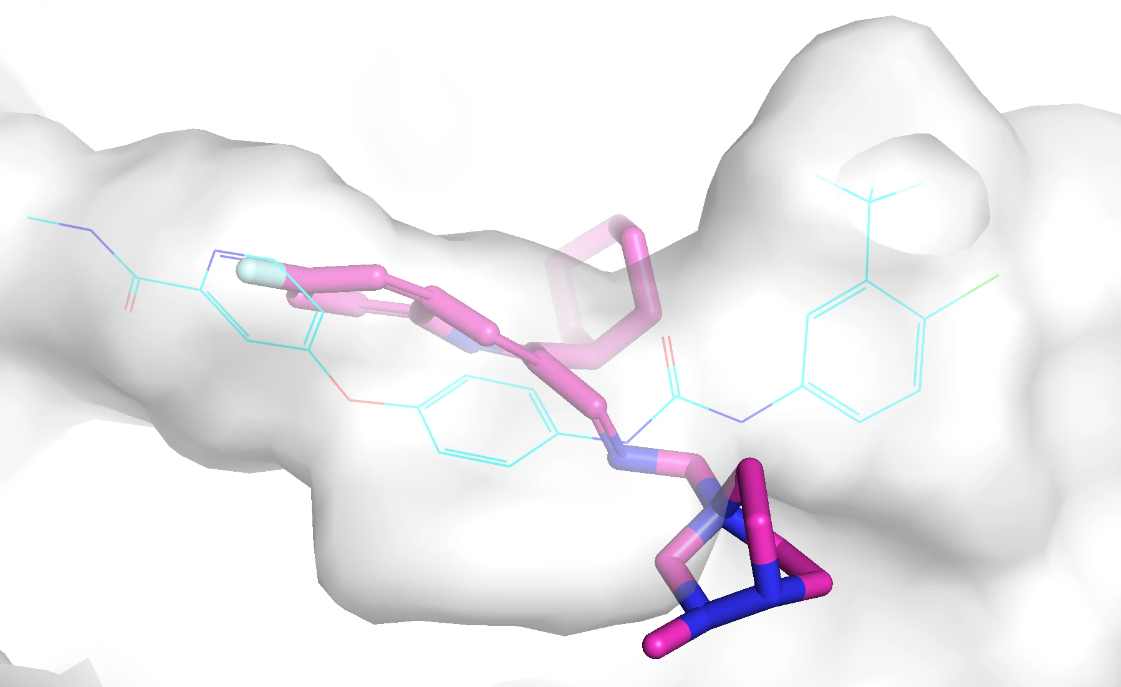
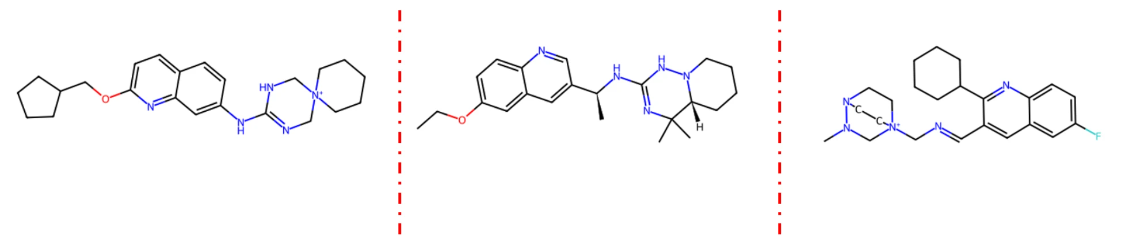
这个三个分子的打分分别是 -10.9,-9.9,-9.8,对应的2D分子结构如下图:
所有生成的分子如下:
CMD-GEN_3WZE
在分子生成过程中,虽然考虑了和给定配体的药效团的匹配程度来选择构象,但是没有考虑分子构象是否和蛋白口袋之间的空间冲突问题。
四、总结
本研究提出的 CMD-GEN 框架通过粗粒化药效团点,将 3D 复合物与 2D 类药分子数据相连接。该模型融合 2D 类药分子与 3D 蛋白-配体复合物信息,显著丰富了生成式训练数据。CMD-GEN 将活性口袋内三维分子生成过程解构为三大模块:口袋条件化三维药效团采样模块、门控条件机制与药效团分子生成模块、基于药效团对齐的分子结合构象生成模块。该方法有效解决了当前生成模型中可能出现的分子构象不稳定性问题。
通过基准实验和真实药物设计任务的综合分析表明:该模型在保持类药属性的同时,能生成具有生物学意义的结合构象;借助门控条件机制进行多参数约束下的微调,实现了分子性质优化。在分子生成效率方面,该模型超越现有最优方法,并凭借点云匹配算法在复杂药物设计任务中表现卓越——例如生成高选择性或多靶点抑制剂。湿实验验证了模型有效性:以 Y5 分子为例,其对PARP1/2 表现出超过 787 倍的选择性。这印证了 CMD-GEN 生成高特异性类药分子的能力,彰显了其在药物研发中的实用价值。CMD-GEN 拓展了药物设计模型的应用边界,为未来研究开辟了重要前景。
尽管 CMD-GEN 在静态全息蛋白结构中表现最优,但静态结构可能因分辨率限制存在偏差,且许多靶标蛋白仅可获得脱辅基构象。未来工作将在 CMD-GEN 框架中融入更多领域知识,例如考虑引入口袋动力学特征。作者相信,这种在有限药物数据条件下将科学原理与模型设计相融合的框架,深刻体现了"科学注入 AI "的理念,必将为开发先进AI驱动的药物设计模型提供新见解与新机遇。
上面是作者在论文最后写的一些讨论总结,作者提供的 V2 版本的模型文件是不完整的,所以只能使用 V1 版本的模型,测试出来 V1 版本生成的分子存在和蛋白口袋的空间冲突问题。V2 版本的性能如何,我们无法测试,所以不得而知。


















 4893
4893




 到【灌水乐园】发言
到【灌水乐园】发言









