




 本文详细介绍了WGCNA在基因共表达网络分析中的应用,包括数据预处理、构建网络、识别modules、筛选相关模块及关键基因,以及数据导出与网络图绘制。通过对小鼠芯片表达谱数据的实例分析,展示了WGCNA在生物信息学研究中的具体操作流程。
本文详细介绍了WGCNA在基因共表达网络分析中的应用,包括数据预处理、构建网络、识别modules、筛选相关模块及关键基因,以及数据导出与网络图绘制。通过对小鼠芯片表达谱数据的实例分析,展示了WGCNA在生物信息学研究中的具体操作流程。
欢迎关注微信公众号《生信修炼手册》!
本文采用WGCNA官网的Tutirial 1的数据,对加权基因共表达网络分析和后续的数据挖掘的具体操作进行梳理
整个分析流程可以分为以下几个步骤
1. 数据预处理
这部分内容包括以下4个部分
- 读取基因表达量数据
- 对样本和基因进行过滤
- 读取样本表型数据
- 可视化样本聚类树和表型数据
官方的示例数据是一个小鼠的芯片表达谱数据,包含了135个雌性小鼠的数据,在提供的表达谱数据中,除了探针ID和样本表达量之外,还有额外的探针注释信息,在读取原始数据时,需要把多余注释信息去除,代码如下
# 读取文件
options(stringsAsFactors = FALSE)
femData = read.csv("LiverFemale3600.csv")
# 去除多余的注释信息列
datExpr0 = as.data.frame(t(femData[, -c(1:8)]))
names(datExpr0) = femData$substanceBXH
rownames(datExpr0) = names(femData)[-c(1:8)]
对于基因的表达量数据,需要进行过滤,对于基因而言,可以过滤缺失值或者低表达的基因,对于样本而言,如果该样本中基因缺失值很多,也需要过滤,WGCNA内置了一个检验基因和样本的函数,通过该函数可以进行一个基本过滤,代码如下
gsg = goodSamplesGenes(datExpr0)
if (!gsg$allOK) {
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
goodSamples和goodGenes就是需要保留的基因和样本。基础过滤之后,还可以看下是否存在离群值的样本,通过样本的聚类树进行判断,代码如下
pdf(file = "sampleClustering.pdf", width = 15, height = 10);
par(cex = 0.6);
plot(sampleTree,
main = "Sample clustering to detect outliers",
sub="", xlab="", cex.lab = 1.5,
cex.axis = 1.5, cex.main = 2)
dev.off()
生成的图片如下
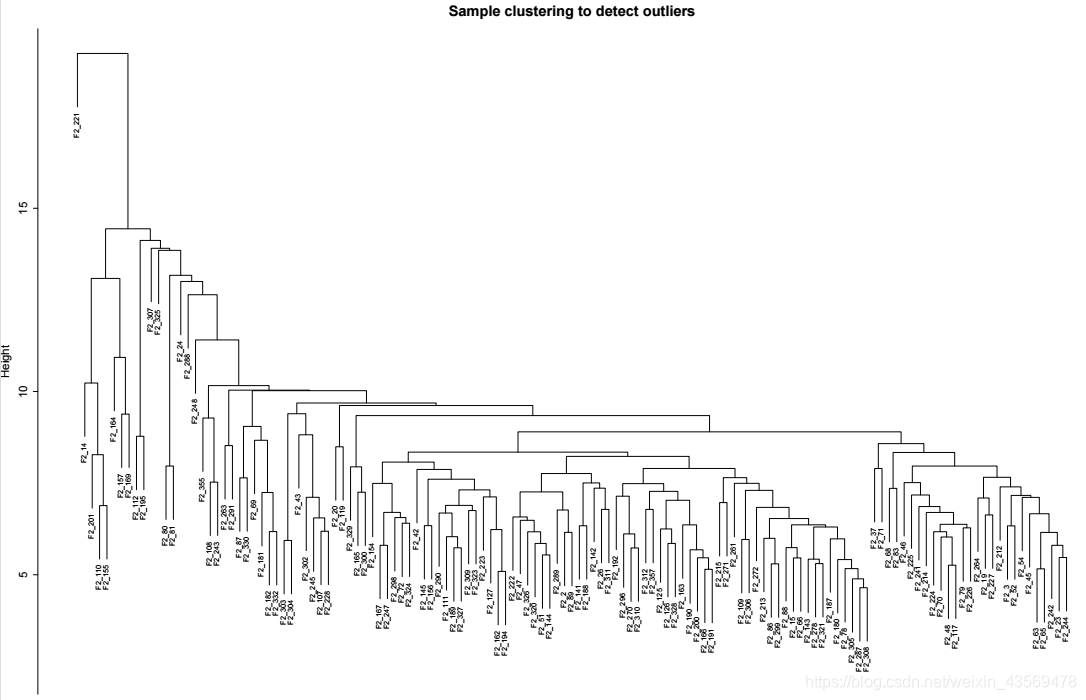
从图上可以看出,F2_221 这个样本和其他样本差距很大,可以将该样本过滤掉。代码如下
clust = cutreeStatic(
sampleTree,
cutHeight = 15,
minSize = 10)
keepSamples = (clust==1)
datExpr = datExpr0[keepSamples, ]
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
表型数据中也包含了不需要的列,而且其样本比表达谱的样本多,需要根据表达谱的样本提取对应的表型数据,代码如下
# 读取文件
traitData = read.csv("ClinicalTraits.csv")
# 删除多余的列
allTraits = traitData[, -c(31, 16)]
allTraits = allTraits[, c(2, 11:36) ]
# 报纸和表达谱的样本一致
femaleSamples = rownames(datExpr)
traitRows = match(femaleSamples, allTraits$Mice)
datTraits = allTraits[traitRows, -1]
rownames(datTraits) = allTraits[traitRows, 1]
表达谱数据和表型数据准备好之后,可以绘制样本聚类树和表型的热图,代码如下
# 由于去除了样本,重新对剩余样本聚类
sampleTree2 = hclust(dist(datExpr), method = "average")
traitColors = numbers2colors(datTraits, signed = FALSE)
plotDendroAndColors(
sampleTree2,
traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
< 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章

















 523
523

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









