




欢迎关注微信公众号《生信修炼手册》!
在解读传统的富集分析结果时,经常会有这样的疑问,一个富集到的通路下,既有上调差异基因,也有下调差异基因,那么这条通路总体的表现形式究竟是怎样呢,是被抑制还是激活?或者更直观点说,这条通路下的基因表达水平在实验处理后是上升了呢,还是下降了呢?
在这里我说下自己的观点,在传统的富集分析时,我们只需要一个差异基因的列表,根本不关心这个差异基因究竟是上调还是下调。这是因为,传统的富集分析根本不需要考虑基因表达量的变化趋势,其算法的核心只关注这些差异基因的分布是否和随机抽样得到的分布一致,即使后期在可视化时,我们在通路图上用不同颜色标记了上下调的基因,但是由于没有采用有效的统计学手段去分析这条通路下所有差异基因的总体变化趋势,这使得传统的富集分析结果无法回答上述的问题。
当然也有人灵光一闪,想出一个解决方案,在进行传统的富集分析时,每次只提取上调或者下调的差异基因来进行分析,由于事先根据表达量变化趋势对差异基因进行了筛选,从而回避了上面的问题。在我个人看来,这样的做法有失偏颇,因为费舍尔精确检验就是想要证明我这个差异基因列表不是随机抽样得到的,而我们事先对差异基因列表的过滤已经对结果的随机性造成了干扰,最后得出的结论其准确性也大大降低。
想象一下,上调基因和下调基因分开富集,然后富集到了同一条通路,这怎么解释?所以在我看来,传统的富集分析只能定位到功能,这些差异基因与哪些功能相关,而不能回答一开始的这个问题。想要回答一开始的这个问题,我们需要GSEA富集方法的结果。
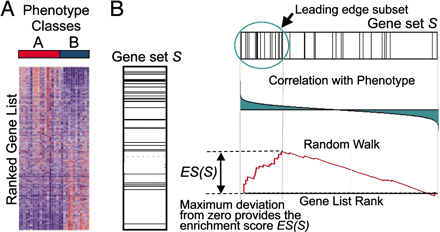
还是这张原理图,GSEA的输入是一个基因表达量矩阵,其中的样本分成了A和B两组,首先对所有基因进行排序,在之前的文章中也有提到排序的标准,这里简单理解就是foldchange, 用来表示基因在两组间表达量的变化趋势。排序之后的基因列表其顶部可以看做是上调的差异基因,其底部是下调的差异基因。
GSEA分析的是一个基因集下的所有基因是否在这个排序列表的顶部或者底部富集,如果在顶部富集,我们可以说,从总体上看,该基因集是上调趋势,反之,如果在底部富集,则是下调趋势。
理解这个观点之后,在来看GSEA富集分析的结果。由于结果很多,所以给出了一个汇总的html页面。
对于富集结果,根据上调还是下调分成了两个部分,对应两个分组,示例如下

在每个组别下富集到的基因集,从总体上看,其表达量在该组中高表达。点击enrichment results in html,可以在网页查看富集的结果,示例如下
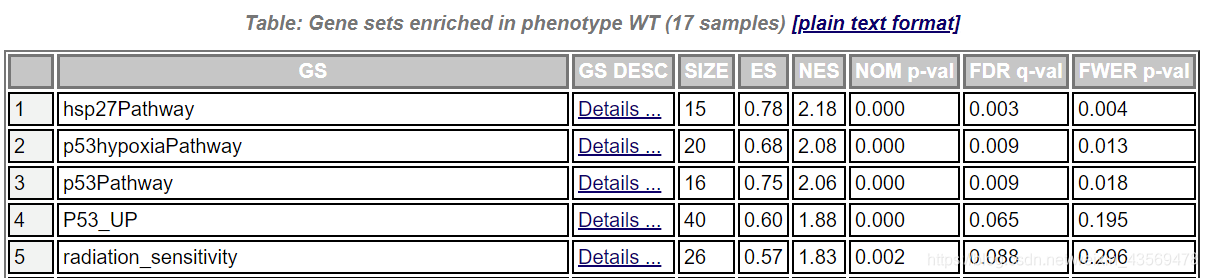
GS为基因集的名字,SIZE代表该基因集下的基因总数,ES代表Enrichment score, NES代表归一化后的Enrichment score, NOM p-val代表pvalue,表征富集结果的可信度,FDR q-val代表qvalue, 是多重假设检验矫正后的p值,注意GSEA采用pvalue < 5%, qvalue < 25% 对结果进行过滤。
点击GS DESC可以跳转到每个基因集详细结果页面,示例如下
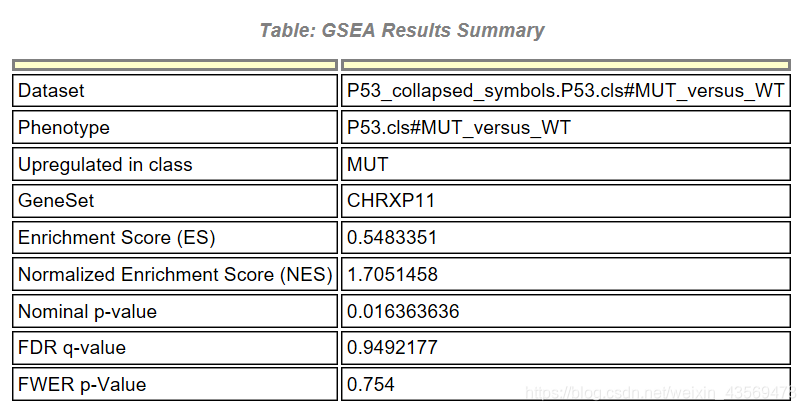
首先是一个汇总的结果,Upregulated in class说明该基因集在MUT这组中高表达,其他信息和之前介绍的一样,除此之外,还有一个详细的表格,示例如下
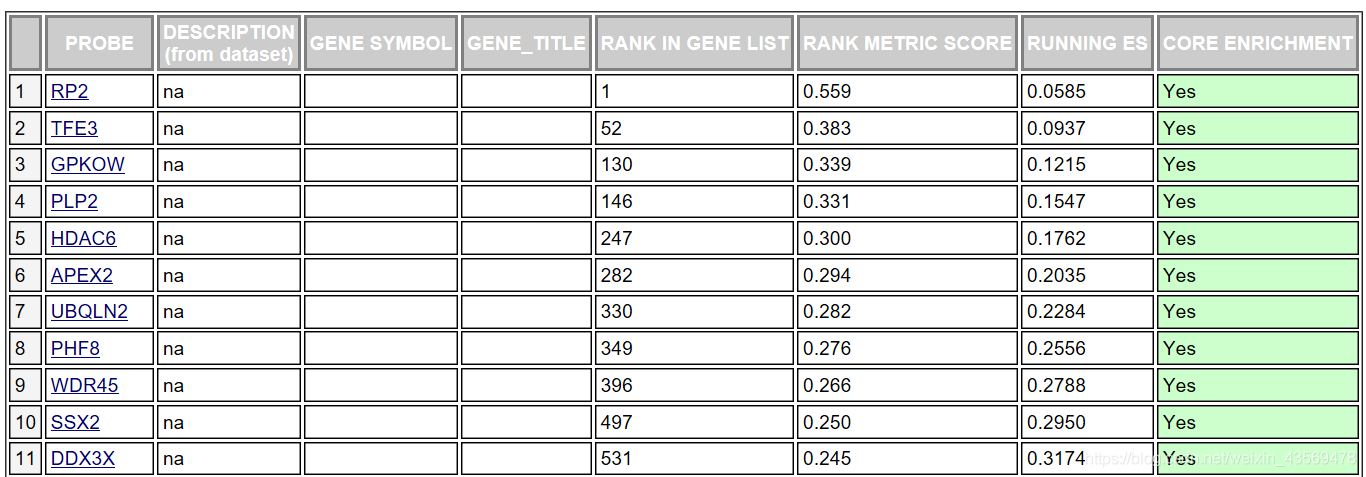
对于该基因集下的每个基因给出了详细的统计信息,RANK IN GENE LIST代表该基因在排序号的列表中的位置, RANK METRIC SCORE代表该基因排序量的值,比如foldchange值,RUNNIG ES代表累计的Enrichment score, CORE ENRICHMENT代表是否属于核心基因,即对该基因集的Enerchment score做出了主要贡献的基因。
这个表格中的数据对应下面这张图
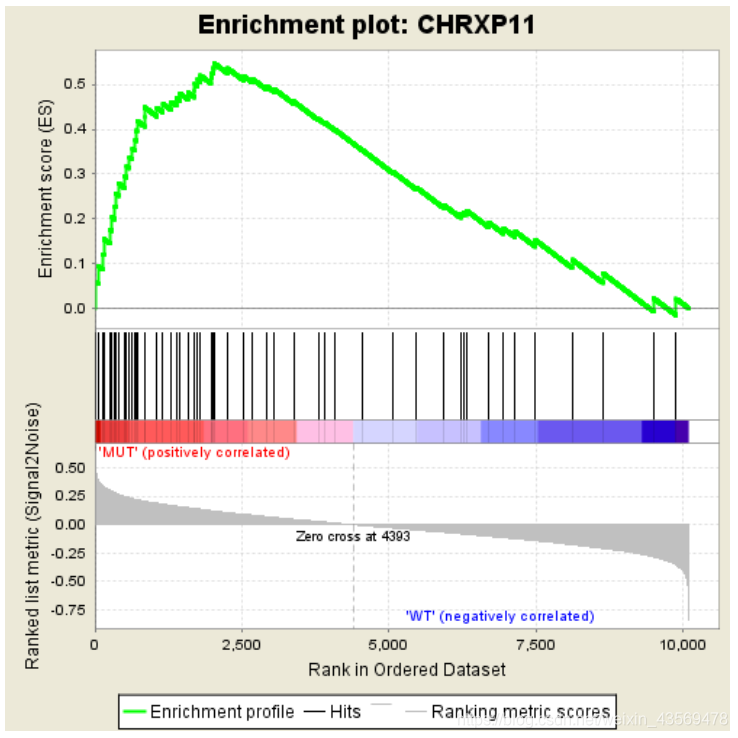
分成3个部分,第一部分为基因Enrichment Score的折线图,横轴为该基因下的每个基因,纵轴为对应的Running ES, 在折线图中有个峰值,该峰值就是这个基因集的Enrichemnt score,峰值之前的基因就是该基因集下的核心基因。
第二部分为hit,用线条标记位于该基因集下的基因,第三部分为所有基因的rank值分布图, 默认采用Signal2Noise算法,对应了纵轴的标题。
从该图中可以看出,这个基因集是在MUT这一组高表达的,下面是一个在另一组组中高表达的示例
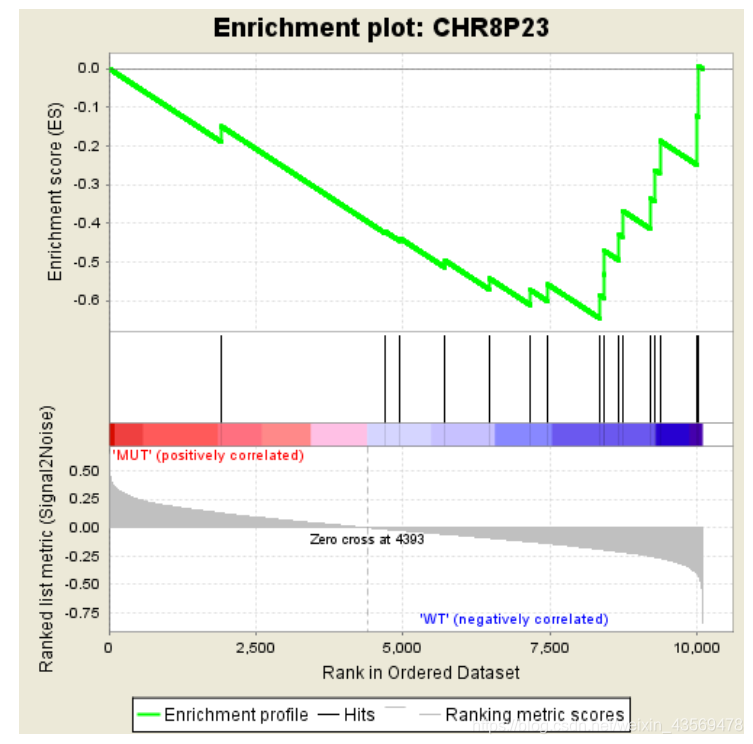
可以看到,其Enrichment score值全部为负数,对应的在其峰值右侧的基因为该基因集下的核心基因。
除此之外,还有一种热图,示例如下
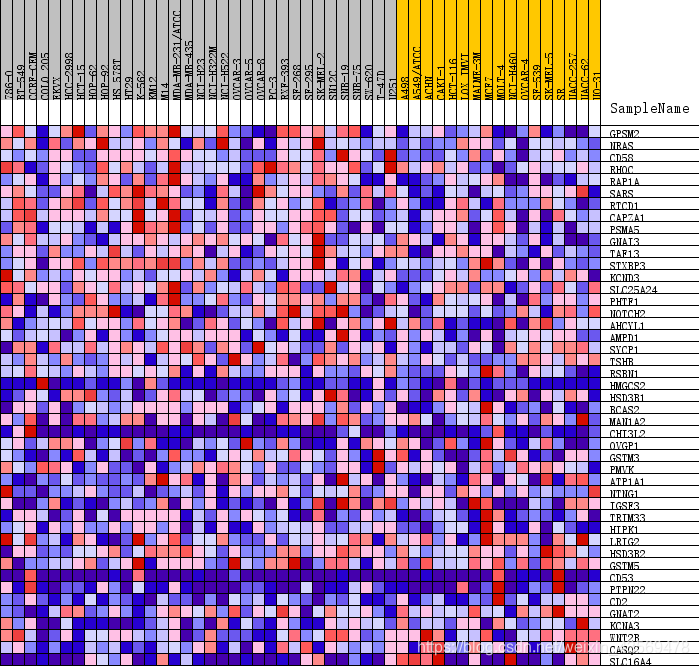
这张热图展示的是位于该基因集下的基因在所有样本中表达量的分布,其中每一列代表一个样本。每一行代表一个基因,基因表达量从低到高,颜色从蓝色过渡到红色。
在总的html页面中,还给出了如下信息
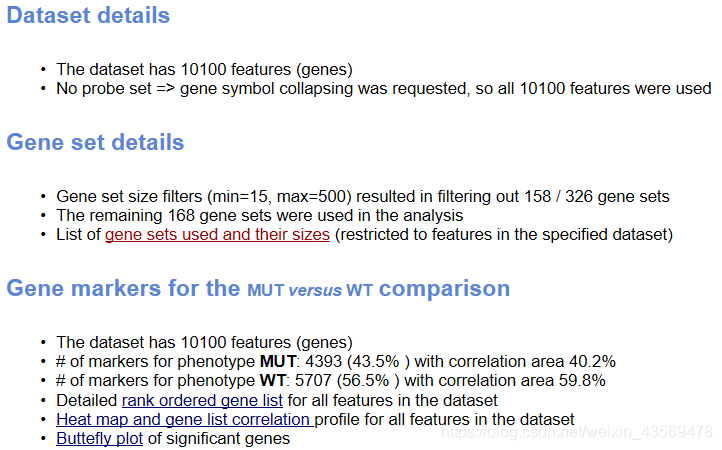
Dataset details给出了基因总数,Gene Set details给出了基因集的信息,注意软件默认根据基因集包含的基因个数是先对基因集进行过滤,最小15个,最大500个基因,过滤掉了158个基因集,剩余的168个基因集用于分析。
Gene markers给出了排序之后的基因列表和对应的统计量rank ordered gene list,根据排序的统计量,将基因分成了两部分,对应在每一组中高表达。heatmap and gene list包含了所有基因表达量的热图和排序值的分布图,示意如下
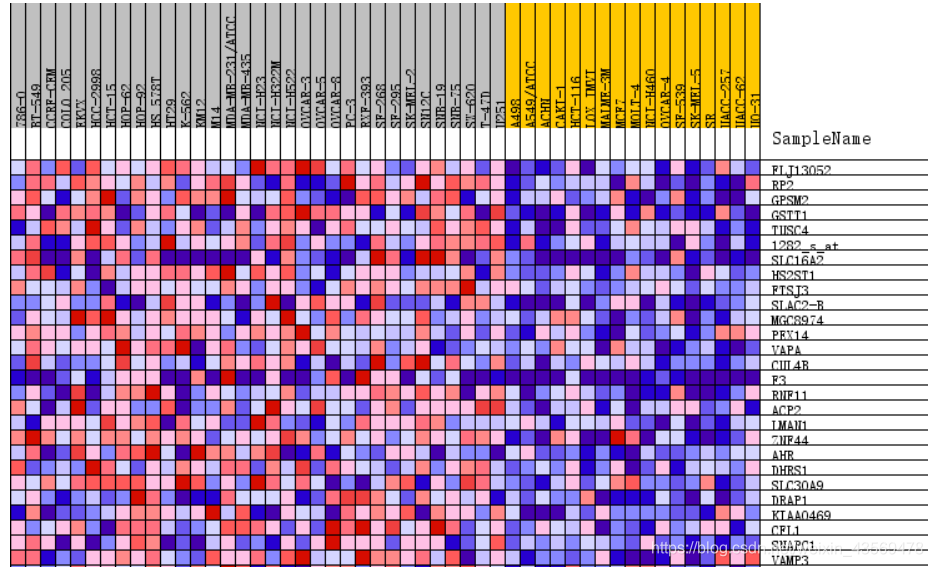
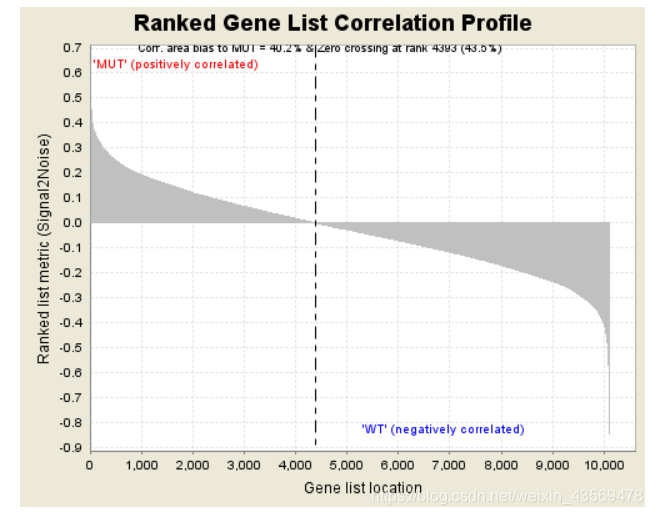
热图由于基因太多,截取了部分,排序值的分布图其实就是每个基因集的Enrichment plot中的第三部分。更多的细节请查阅官方文档。
扫描关注微信号,更多精彩内容等着你!


















 175
175

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









