







简 介
群体分层长期以来被认为是遗传关联研究中的一个混淆因素。根据多基因座基因型数据估算的祖先可用于对种群分层进行统计校正。一种流行的估算祖先的技术是基于模型的方法,它体现在广泛应用的程序结构中。在程序 EIGENSTRAT 中实现的另一种方法依赖于主成分分析,而不是基于模型的估计,并且不直接提供混合物分数。EIGENSTRAT 已获得的普及,部分原因是由于其显著的速度相比结构。我们提出了一种新的算法和程序,ADMIXTURE用于基于模型的估计无亲缘关系个体的祖先。ADMIXTURE 采用嵌入结构的似然模型。然而,ADMIXTURE 运行速度快得多,在几分钟内解决问题,需要结构小时。在我们的许多实验中,我们发现 ADMIXTURE 几乎和 EIGENSTRAT 一样快。ADMIXTURE 的运行时改进依赖于使用顺序二次规划进行块更新的快速块松弛方案,以及新颖的准牛顿收敛加速。我们的算法也运行得更快,并且比合并在程序FRAPPE中的期望最大化(EM)算法的实现具有更高的精度。我们的模拟表明,ADMIXTURE 对潜在外加剂系数和祖先等位基因频率的最大似然估计与结构的贝叶斯估计一样准确。在真实世界的数据集上,ADMIXTURE 的估计可以直接与 structure 和 EIGENSTRAT 的估计进行比较。综上所述,我们的结果表明 ADMIXTURE 的计算速度打开了在基于模型的祖先估计中使用更大标记集的可能性,并且其估计适合用于校正关联研究中的人口分层。

软件包安装
软件包下载链接:
https://dalexander.github.io/admixture/binaries/admixture_linux-1.3.0.tar.gz
测试数据下载链接:
https://dalexander.github.io/admixture/hapmap3-files.tar.gz
解压之后进入文件夹里面找到 admixture,运行即可。
$tar -xvf hapmap3-files.tar.gz
$cd dist/admixture_linux-1.3.0/
./admixture --help
**** ADMIXTURE Version 1.3.0 ****
**** Copyright 2008-2015 ****
**** David Alexander, Suyash Shringarpure, ****
**** John Novembre, Ken Lange ****
**** ****
**** Please cite our paper! ****
**** Information at www.genetics.ucla.edu/software/admixture ****
ADMIXTURE basic usage: (see manual for complete reference)
% admixture [options] inputFile K
where:
K is the number of populations; and
inputFile may be:
- a PLINK .bed file
- a PLINK "12" coded .ped file
Output will be in files inputBasename.K.Q, inputBasename.K.P
General options:
-jX : do computation on X threads
--seed=X : use random seed X for initialization
Algorithm options:
-m=
--method=[em|block] : set method. block is default
-a=
--acceleration=none |
sqs<X> |
qn<X> : set acceleration
Convergence criteria:
-C=X : set major convergence criterion (for point estimation)
-c=x : set minor convergence criterion (for bootstrap and CV reestimates)
Bootstrap standard errors:
-B[X] : do bootstrapping [with X replicates]数据准备
解压测试数据集,可以看到里面包含了三个文件,这三个文件都是通过plink软件生产而来。
$tar -xvf hapmap3-files.tar.gz
$ls
hapmap3.bed hapmap3.bim hapmap3.fam可以通过PLINK生产 .ped 12"编码文件,该文件由以下命令生成
plink --file hapmap --recode12 --out hapmap实例操作
已知群体结构
你相信样本中的个体的祖先来自三个祖先人群,然后像这样进行混合:
./admixture hapmap3.bed 3若是 .ped 执行下面的命令,生成两个文件.Q和.P:
$./admixture hapmap3.ped 3
## 产生文件
hapmap3.3.Q hapmap3.3.P如果还需要标准错误,那么应该使用命令,可以看到结果文件多了 .Q_se。
./admixture -B hapmap3.ped 3
## 生成文件
hapmap3.3.Q hapmap3.3.P hapmap3.3.Q_se如果您确实需要分析所有标记,或者由于其他原因(非常大的K,广泛的交叉验证或引导),您可以考虑在多线程模式下运行 ADMIXTURE。例如,如果您的计算机有四个处理器,您可以使用这样的命令:
./admixture hapmap3.bed 3 -j4未知结构如何选择K的正确值?
使用 ADMIXTURE 的交叉验证程序。与其他K值相比,良好的K值将表现出较低的交叉验证误差。只需在ADMIXTURE 命令行中添加——cv标志即可启用交叉验证。在这个默认设置中,交叉验证过程将执行5倍CV,例如,使用——cv =10,可以得到10倍CV。在输出中报告交叉验证错误。
$for K in 1 2 3 4 5 6 7 8 9 10 ; do ./dist/admixture_linux-1.3.0/admixture --cv hapmap3.bed 10 | tee log10.out; done
$grep -h CV log*.out > CVerror.txt
CV error (K=10): 0.53864
CV error (K=1): 0.55248
CV error (K=2): 0.48190
CV error (K=3): 0.47835
CV error (K=4): 0.48236
CV error (K=5): 0.49001
CV error (K=6): 0.50069
CV error (K=7): 0.51484
CV error (K=8): 0.51761
CV error (K=9): 0.52387群体结构可视化
将CV结果复制粘贴至文本中中,ggplot2 绘制折线图。图中可看出最佳分群数为K=3。
CVerror = read.table("CVerror.txt")
CVerror[, 3]
## [1] "(K=10):" "(K=1):" "(K=2):" "(K=3):" "(K=4):" "(K=5):" "(K=6):"
## [8] "(K=7):" "(K=8):" "(K=9):"
library(stringr)
data = data.frame(K = str_extract(CVerror[, 3], "([:digit:]{1,2})"), Error = CVerror[,
4])
data$K = as.integer(data$K)
library(ggplot2)
ggplot(data, aes(x = K, y = Error)) + geom_line() + geom_point() + ylab("Cross−validation error") +
scale_x_continuous(breaks = seq(1, 10, 1))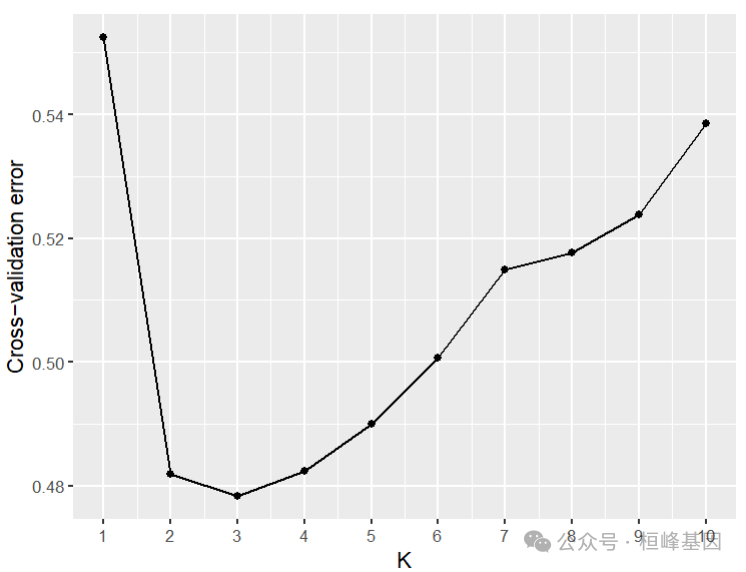
如何画出Q的估计值呢?
Q估计值作为一个简单的矩阵输出,要制作堆叠的条形图,使用barplot命令。
tbl = read.table("hapmap3.3.Q")
colnames(tbl) = c("Q1", "Q2", "Q3")
barplot(t(as.matrix(tbl)), col = rainbow(3), xlab = "Individual #", ylab = "Ancestry",
border = NA)
legend(x = 5, y = -0.01, pch = 15, colnames(tbl), col = rainbow(3), bty = "n", horiz = TRUE,
xpd = TRUE)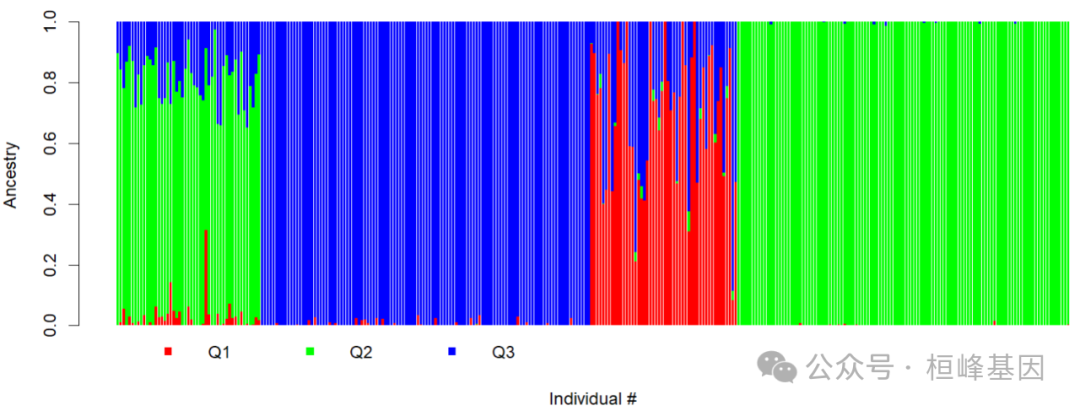
Reference
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009 Sep;19(9):1655-64.
基因组生信分析教程
DNA 1. Germline Mutation Vs. Somatic Mutation 傻傻分不清楚
DNA 2. SCI 文章中基因组变异分析神器之 maftools
DNA 3. SCI 文章中基因组变异分析神器之 maftools
DNA 4. SCI 文章中基因组的突变信号(maftools)
DNA 6. 基因组变异之绘制精美瀑布图(ComplexHeatmap)
DNA 7. 基因组拷贝数变异分析及可视化 (GISTIC2.0)
DNA 8. 癌症的突变异质性及寻找新的癌症驱动基因(MutSigCV)
DNA 10. 识别癌症驱动基因 (OncodriveCLUST)
DNA 11. 识别肿瘤蛋白质三维结构上突变热点(HotSpot3D)
DNA 12. SCI 文章绘图之全基因组关联分析可视化(GWAS)
DNA 14. SCI 文章肿瘤微卫星不稳定性计算方法(MSI)
DNA 15. SCI 文章肿瘤微卫星不稳定性之 MSIsensor 系列软件
DNA 16. SCI 文章研究表型与基因型之间的关系工具(TASSEL)

















 3384
3384

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









