









MAHAKIL: Diversity Based Oversampling Approach to Alleviate the Class Imbalance Issue in Software Defect Prediction论文解读
摘要
高度不平衡的数据通常会使准确预测变得困难。不幸的是,软件缺陷数据集往往有缺陷的模块比无缺陷的模块少。合成过采样方法通过创建新的在训练模型之前平衡类分布的少数有缺陷的模块。尽管取得了成功这些方法主要导致过度泛化(误报率高)并生成近乎重复的数据实例(不太多样化的数据)。在这项研究中,我们介绍了 MAHAKIL,一种新颖有效的软件缺陷合成过采样方法基于遗传的染色体理论的数据集。利用这一理论,MAHAKIL 将两个不同的子类解释为父母并生成一个新实例,该实例从每个父母那里继承了不同的特征,并有助于数据的多样性分配。我们将 MAHAKIL 与 SMOTE、Borderline-SMOTE、ADASYN、Random Oversampling 和 No使用来自 PROMISE 存储库的 20 个版本的缺陷数据集和五个预测模型的抽样方法。我们的实验表明 MAHAKIL 提高了所有模型的预测性能,并实现了比其他过采样方法,基于 Brunner 的统计显着性检验和 Cliff 的效应大小。因此,MAHAKIL 是强烈推荐作为基于高度不平衡数据集的缺陷预测模型的有效替代方案。
步骤一、样本多样性测量
定义两个样本示例
x
,
y
x,y
x,y如下
x
=
(
x
1
,
x
2
,
…
,
x
n
)
T
x=(x_1,x_2,\dots,x_n)^T
x=(x1,x2,…,xn)T
y
=
(
y
1
,
y
2
,
…
,
y
n
)
T
y=(y_1,y_2,\dots,y_n)^T
y=(y1,y2,…,yn)T
两个示例的马哈拉诺比斯距离就定义为
d
(
x
,
y
)
=
(
x
−
y
)
T
S
−
1
(
x
−
y
)
d(x,y)=\sqrt{(x-y)^TS^{-1}(x-y)}
d(x,y)=(x−y)TS−1(x−y)
其中
S
−
1
S^{-1}
S−1是样本空间上的协方差,下面举个计算协方差的简单的例子
我们现在有三个样本
x
1
=
(
0
,
1
,
3
)
、
x
2
=
(
2
,
3
,
0
)
、
x
3
=
(
4
,
2
,
3
)
x_1=(0,1,3)、x_2=(2,3,0)、x_3=(4,2,3)
x1=(0,1,3)、x2=(2,3,0)、x3=(4,2,3)将其写成矩阵的形式
A
=
(
0
1
3
2
3
0
4
2
3
)
A=\begin{pmatrix} 0 & 1 & 3 \\ 2 & 3 & 0 \\ 4 & 2 & 3 \\ \end{pmatrix}
A=⎝⎛024132303⎠⎞
计算公式:
∑
i
j
=
(
第
i
列
−
第
i
列
的
均
值
)
T
×
(
第
j
列
−
第
j
列
的
均
值
)
样
本
数
−
1
\sum_{ij}= \frac{(第i列 - 第i列的均值)^T\times(第j列 - 第j列的均值)}{样本数 - 1}
∑ij=样本数−1(第i列−第i列的均值)T×(第j列−第j列的均值)
∑ 11 = ( − 2 , 0 , 2 ) T × ( − 2 , 0 , 2 ) 3 − 1 = 4 , ∑ 12 = ( − 2 , 0 , 2 ) T × ( − 1 , 1 , 0 ) 3 − 1 = 1 … \sum_{11}=\frac{(-2,0,2)^T\times(-2,0,2)}{3 - 1}=4, \sum_{12}=\frac{(-2,0,2)^T\times(-1,1,0)}{3 - 1}=1\dots ∑11=3−1(−2,0,2)T×(−2,0,2)=4,∑12=3−1(−2,0,2)T×(−1,1,0)=1…最后得到的每一个 ∑ i j \sum_{ij} ∑ij组成的矩阵 S S S就是协方差矩阵
步骤二、数据划分和配对
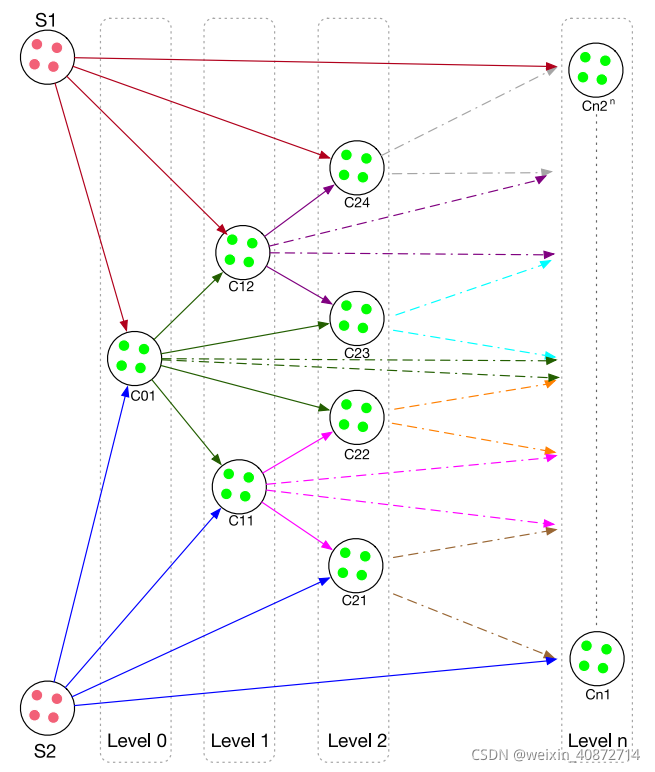
基于染色体遗传理论中建立的双亲本染色体相等的概念,论文根据较小的马氏距离和较大的马氏距离将数据样本划分或分离到两个盒子中。论文提到经过多次模拟实验,表明这种方式可以输出最佳和最合适的合成数据。论文中将样本按照马氏距离降序排列,以中间值为分界将整个样本的数据划分为两个相同的部分,其中大于等于中间值的划分为一类。配对是按照顺序完成的,即两个盒子的第一个实例都给一个一样的标签,将其标记为一对,依次按序配对。
步骤三、合成样本的生成
在最后阶段,通过聚合并计算来自每个分区的两个配对实例之间的平均值来生成合成样本,然后将其添加到原始数据中,基于在上一阶段中创建的两个盒子,生成的结果实例将明显是唯一的,并且与来自这两个盒子的父实例相关。使用这种方法的好处是,生成的合成样品在大多数区域不重叠,其分布比较平均。合成样本技术常用的是k-NN算法,由基于k-NN的技术生成的合成样本倾向于聚类在少数类样本中的特定群体中,只能为训练过的分类器提供有限的信息。而相比于k-NN算法,由MAHAKIL生成的合成样本均匀分布,使用了两个盒子中所有可能的样本,从而为分类器提供更多的信息。
分析算法

算法的输入是整个数据集
N
N
N和过采样需要达到的平衡比例
P
P
P or
P
f
p
Pfp
Pfp
step 1: 将数据集
N
N
N划分为两个数据集
N
m
i
n
N_{min}
Nmin和
N
m
a
j
N_{maj}
Nmaj分别代表小样本数据集和大样本数据集
step 2: 使用
P
f
p
Pfp
Pfp 计算出
T
T
T ,计算出
k
=
a
r
r
a
y
l
e
n
g
t
h
(
N
m
i
n
)
k=arraylength(N_{min})
k=arraylength(Nmin)。
T
T
T表示达到平衡比例
P
f
p
Pfp
Pfp需要生成的合成样本数量,
k
k
k表示
N
m
i
n
N_{min}
Nmin的元素个数。
step 3: 初始化数组
X
n
e
w
X_{new}
Xnew ,用来存储合成样本。
step 4: 初始化整型变量
X
n
e
w
c
h
k
i
X_{newchki}
Xnewchki,表示合成样本的个数。
step 5: 计算
N
m
i
n
N_{min}
Nmin中每个样本的马氏距离
D
2
D^2
D2,
D
2
=
(
x
−
μ
)
′
∑
−
1
(
x
−
μ
)
D^2=(x-\mu)^\prime\sum^{-1}(x-\mu)
D2=(x−μ)′∑−1(x−μ)。其中
μ
\mu
μ是整个样本的均值,
∑
\sum
∑是协方差矩阵。
μ
=
1
n
∑
i
=
1
n
x
i
\mu=\frac{1}{n}\sum_{i=1}^{n}x_i
μ=n1∑i=1nxi
step 6: 将每个样本的马氏距离和他所对应的样本按照降序存储到
N
m
i
n
d
i
s
t
N_{mindist}
Nmindist中。
step 7: 找到样本的中间值的序号,用
N
m
i
d
=
k
/
2
N_{mid}=k/2
Nmid=k/2表示。
step 8: 通过
N
m
i
d
N_{mid}
Nmid和
N
m
i
n
d
i
s
t
N_{mindist}
Nmindist将
N
m
i
n
N_{min}
Nmin分为两个集合
N
b
i
n
1
=
{
y
1
,
y
2
,
…
,
y
m
i
d
}
N_{bin1}=\lbrace y_1,y_2,\dots,y_{mid}\rbrace
Nbin1={y1,y2,…,ymid} 和
N
b
i
n
2
=
{
y
m
i
n
d
+
1
,
y
m
i
n
d
+
2
,
…
,
y
k
}
N_{bin2} = \lbrace y_{mind+1},y_{mind+2},\dots,y_{k}\rbrace
Nbin2={ymind+1,ymind+2,…,yk}。
step 9: 对于每一个
y
i
∈
N
b
i
n
1
y_i\in N_{bin1}
yi∈Nbin1和
y
i
∈
N
b
i
n
2
y_i\in N_{bin2}
yi∈Nbin2,按顺序分配一个唯一的标签(
l
i
l_i
li)
i
=
1
…
…
m
i
d
i = 1\dots\dots mid
i=1……mid。
step 10-12: 使用循环从1到mid 选取下标相同的两个样本
y
a
,
y
b
y_a,y_b
ya,yb,取其平均
x
=
a
v
e
r
a
g
e
(
y
a
,
y
b
)
x = average(y_a,y_b)
x=average(ya,yb),将
x
x
x添加到
X
n
e
w
X_{new}
Xnew并将
X
n
e
w
c
h
k
X_{newchk}
Xnewchk的值加1。
step 13 : if
X
n
e
w
c
h
k
<
T
X_{newchk} < T
Xnewchk<T,表示合成样本数量不够,那么将
X
n
e
w
X_{new}
Xnew中新合成的样本分别和
N
b
i
n
1
,
N
b
i
n
2
N_{bin1},N_{bin2}
Nbin1,Nbin2配对,重复10-12的步骤,获得
X
n
e
w
[
j
]
(
j
=
1
,
2
)
X_{new[j]}(j=1,2)
Xnew[j](j=1,2)。如果
X
n
e
w
c
h
k
<
T
X_{newchk} < T
Xnewchk<T 依然成立,则将
X
n
e
w
[
j
]
X_{new[j]}
Xnew[j] 与其父节点与它的直接父集配对,然后与直接父集的前任集合配对,并在后续回合中重复步骤10到12。
step 14: if
X
n
e
w
c
h
k
≥
T
X_{newchk} \geq T
Xnewchk≥T ,将
X
n
e
w
X_{new}
Xnew中的所有样本加上小样本的标签添加到
N
N
N中
图解算法过程
小组讨论的想法
- 马氏距离的 μ \mu μ到底是整个样本空间的均值还是单个样本的均值
- 论文使用马氏距离求样本多样性,其目的就是要找到尽可能不同的样本作为父类生成样本,那么能不能想出其他的方法呢?例如使用聚类的方式。
- 子类的生成方法也很简单粗暴,直接取平均。那么对于特征的不同维度可以采取不同的方式吗?例如对特征加权。
- 生成的子类是更像父类中的那一个呢?这可能需要运用到统计学习的方法
- 文章的思想对许多的不平衡的数据都是可以通用的,只是对于不同的数据集采用什么方法才能得到更好的效果

















 505
505

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









