




 本文详细介绍了ChIA-PET技术,包括背景、测序原理、步骤和数据处理。ChIA-PET利用双交联固定DNA-蛋白质复合物,通过超声破碎和目标蛋白抗体富集,进行染色质相互作用分析。数据处理涉及连接子过滤、PET映射、冗余去除等7个步骤。此外,文章还提到了ChIA-PET Tool、Mango、MICC和ChIA-PET2等数据分析方法,并比较了它们的性能。
本文详细介绍了ChIA-PET技术,包括背景、测序原理、步骤和数据处理。ChIA-PET利用双交联固定DNA-蛋白质复合物,通过超声破碎和目标蛋白抗体富集,进行染色质相互作用分析。数据处理涉及连接子过滤、PET映射、冗余去除等7个步骤。此外,文章还提到了ChIA-PET Tool、Mango、MICC和ChIA-PET2等数据分析方法,并比较了它们的性能。
一、背景介绍
利用成对末端标记序列(ChIA-PET)进行染色质相互作用分析是一种将功能染色质结构转化为数百万个短标记序列的方法
配对末端标签测序ChIA-PET技术应用了PET测序技术的基本原理。PET测序技术的独特之处在于构建配对末端测序模板。从目的DNA片段两个末端提取短标签并将它们配对构成一个PET,对PET进行高通量测序,利用标签序列定位目的DNA片段在基因组中的位置。为了获得短标签,将目的DNA片段两个末端分别与设计好的半连接子(linker)相连。半连接子为具有MmeI限制性内切酶识别位点的寡聚核苷酸序列,而MmeI限制性内切酶能够水解其识别位点下游18/20个碱基对。连接子连接后将DNA片段两个末端配对,随后加入MmeI限制性内切酶,便可得到“标签-连接子-标签(tag-linker-tag)”结构。

二、测序原理
1, 用甲醛铰链DNA和蛋白,目的是让可能存在相互影响的DNA片段连接;
2, 用超声等方法将DNA片段化;
3, 用特异性蛋白抗体富集DNA和蛋白复合物;
4, 在DNA片段末端加上包含MmeI位点的生物素化寡核苷酸linker;
5, 连接linker,从而形成连接目的DNA片段的桥梁;
6, 用限制性内切酶消化得到DNA片段,去除蛋白质;
7, 固定化PET序列;
8,上机测序。
三、测序
1、先乙二醇、再用甲醛进行双交联,固定DNA-蛋白质复合物。细胞溶解去除胞质蛋白
2、使用超声波破碎,染色质通常应该打断为200bp到600bp的DNA片段,其平均长度为500bp。
short-read ChIA-PET: read长度为36bp,测序的DNA片段长度为20bp左右
long-read ChIA-PET: 平均长度为300bp(Tangetal.,2015)
3、加入目标蛋白抗体,富集含有目标蛋白的DNA-蛋白质复合物。
为了区分来自不同蛋白交联区域的序列,ChIA-PET引入了双末端标签(Paired-End Tags)标记。双末端标签的目的在于,区分连接之后的DNA序列是否来自同一个蛋白结合的序列。

4-8.1 short-read ChIA-PET:
A、B两种linker的序列中包含一个指定位置上的短标签(由红色标注)
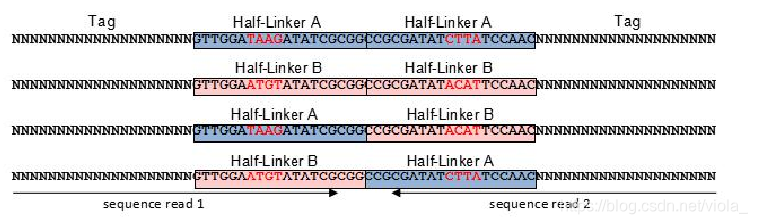
- 两种half-linkerA和B分别加到两等份样品中用于linker连接。
- 将这两等份混合时,两个half-linker连接在一起构成了全长的linker序列。
- half-linker与ChIP-DNA结合的一端中包含MmeⅠ酶的一段识别序列,以实现第三步片段化
- 使用MmeⅠ向ChIP-DNA方向切割20bp,以此得到tag-linker-tag格式的PETs(Paired-EndTags), Tag指的是使用MmeⅠ酶切下来的ChIP-DNA片段
- 每对tags经过PCR扩增
- 测序分析
交联-染色质碎裂-染色质沉淀-两份单独的等分试样中的接头连接-邻近结扎-限制性内切-PET测序
- 如果测序得到的结果中,两段序列的linker都带有相同的短标签,则说明他们来自同一个蛋白结合的序列,存在互相作用,是我们想要得到的序列;
- 相反,如果两段序列的linker带有不同的短标签(chimeric PET),则说明他们来自于不同的蛋白交联,应该从我们的数据分析中被排除。
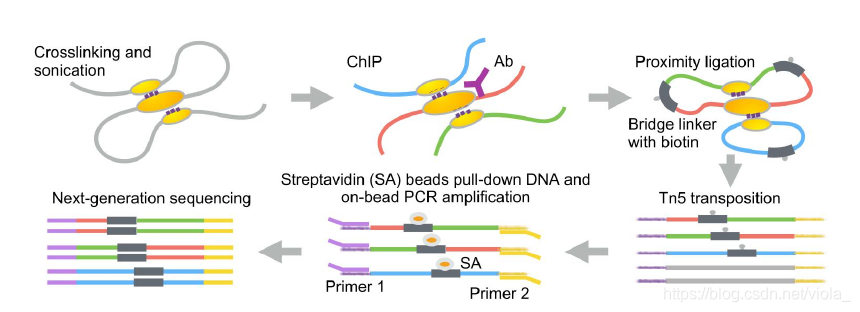
4-8.2 long-read ChIA-PET:
- 加入一条生物素标记的bridge linker序列进行邻近连接
- 使用Tn5转座酶将连接产物片段化,同时加上测序接头
- PCR扩增
- 测序
交联和超声-生物素标记-邻近结扎-Tn5转座酶将连接产物片段化,同时加上测序接头- PCR 扩增-测序
primer(引物):引物是人工合成的两段寡核苷酸序列,一个引物与目的基因一端的一条DNA模板链互补,另一个引物与目的基因另一端的另一条DNA模板链互补
- 长度:18-27bp
- 功能:PCR扩增,影响PCR特异性
四、数据处理
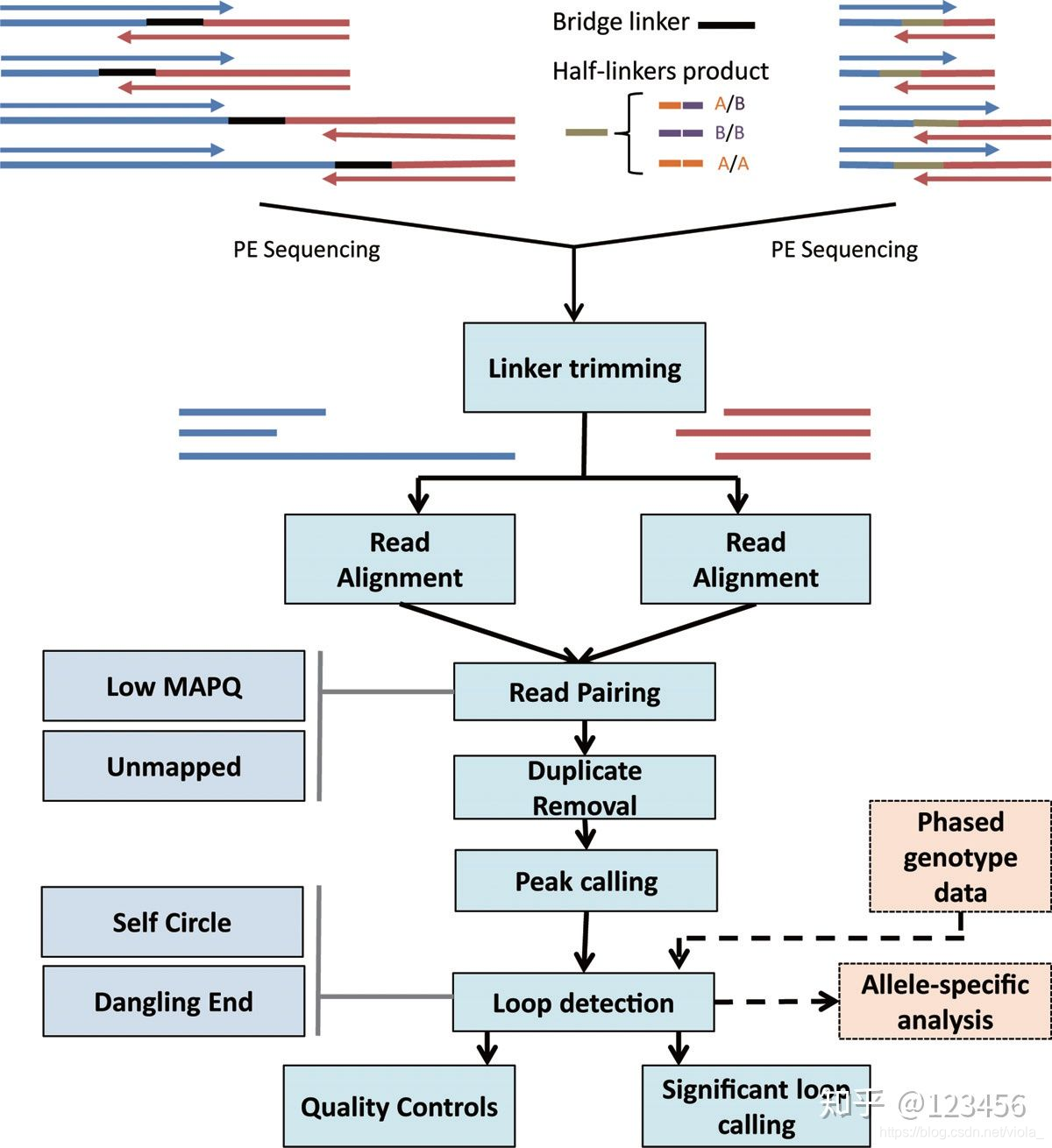
对端测序的结果存储在2个fastq文件中,可以使用ChIA-PET工具(ChIA-PET2)或其他方法进行处理。
ChIA-PET数据处理有7个步骤
1、连接子过滤
- 连接体将与参考半连接体核苷酸序列对齐。除标签序列外,有2种半连接体,分别为A和B,有相同的核苷酸。据连接体的组成将PET分为2类:相同的连接体(AA或BB)和不同的连接体(AB或BA),然后将连接体从原始测序片段中排除,并保留剩余的DNA片段以供进一步分析
2、PET映射
- 用BWA、Bowtie、Batmis或其他绘图工具将短DNA序列与参考基因组对齐
3、冗余去除
- 使用samtools和bedtools过滤掉冗余和低质量的映射序列
4、自连和互连PET分类
- 自连PET:从两端循环的单个DNA片段的测序片段,并在同一染色体上的短距离内映射到基因组。
- 互连PET:来自不同DNA片段的测序片段,通常2个标签位于不同染色体中或长距离位于同一染色体中。
5、结合位点分析自连Pet
- 用自连PET来确定基因组上的蛋白质结合位点
6、用互连Pet进行染色质相互作用分析
- 互连PET可以通过聚类来预测染色质相互作用
7、染色质相互作用数据的可视化
- 构建ChIA-PET浏览器来报告数据并可视化结合位点以及交互集群
五、ChIA-PET数据分析方法
1、ChIA-PET Tool介绍
能够产生准确的交互,但是软件只执行ChIA-PET数据分析、交互评分中的最后一步。软件包不公开。
2、Mango介绍
Mango将基因组位点间相互作用的可能性作为距离和峰深的函数进行建模,并使用该模型为相互作用分配统计置信度。值得注意的是,Mango用一种简单而健壮的贝叶斯方法取代了计算上昂贵的距离匹配重布线方法。
由于使用方便和准确性的提高,Mango将通过对ChIA-PET数据集的分析,大幅提升揭示三维染色质结构特征和功能的能力。同时也纠正了非特定的相互作用,可以作为一个基因组接近和峰深的函数。
Mango被设计成所有的研究人员都可以使用,易安装,只需一个命令就可以完成从fastq到交互的所有步骤。
3、 MICC介绍
MICC,一种易于使用的R包,用于处理ChIA-PET数据。MICC旨在以高灵敏度检测染色质相互作用,同时将错误发现率(FDR)控制在合理水平。 MICC的输入是源自ChIA-PET数据的原始PET簇。 MICC的最终输出包括:将PET簇描述为真实相互作用簇的后验概率列表和相应的FDR。在不同数据集的相同FDR上,MICC总能检测到比ChIA-PET工具和ChiaSig更多的相互作用。此外,MICC检测到的相互作用在生物学重复之间也更加一致。
3、ChIA-PET2介绍
ChIA-PET2能够分析不同类型的ChIA-PET数据从原始的测序读段,形成染色质互作环(chromatin loops)的有效便捷的工具。尽管目前已有很多用于分析ChIA-PET数据的工具,例如,ChIA-PET Tool,ChiaSig,MICC,Mango和3CPET,但ChIA-PET2有其自身的优势

ChIA-PET2整合了ChIA-PET数据分析的所有步骤,包括linker trimming,read alignment,duplicate removal,peak calling和chromatin loop calling。
4、各种工具之间的性能比较
- Mango仅依赖4个广泛使用且易于安装的软件包。
- MICC,从ChIA-PET的数据中检测显著染色质相互作用。
- 与ChIA-PET Tool相比,MICC使用较低深度的测序库恢复了较高深度测序库中检测到的交互作用的显著比例。还为PET集群提供了更一致的排序,从而可以提高实验复制之间的再现性。
- ChIA-PET2支持不同类型的ChIA-PET技术产生的ChIA-PET数据,并针对不同CHIA-PET分析步骤提供质量控制。将ChIA-PET2应用于不同的ChIA-PET数据集,在处理原始的ChIA-PET数据时,ChIA-PET2都有良好的性能、稳定性以及易操作性
参考:
https://www.bilibili.com/video/av970780224?p=61

















 327
327

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









