






最近在读一篇文献的时候,了解到一个R包-scPred。有什么用呢,且听我娓娓道来。首先平时我们拿到单细胞数据集,经过seurat标准流程,降维聚类,然后根据已知的marker基因对细胞进行注释,这个过程的降维分群是一个无监督的过程。而scPred结合机器学习算法,提供的是一种有监督的细胞注释分群。有什么用呢?假设这样一个场景,我们预先对某一个数据进行了完美的分群注释,尤其是细胞亚群,当我们要对另外一个数据集进行注释的时候,就可以将之前注释好的数据当作training数据,当前数据作为test数据,根据之前的注释对当前数据集进行分类。还有,做类器官发育的时候,需要要发育阶段的细胞进行分群注释,我们这个时候可以参考相同物种的正常发育顺序的单细胞数据集,对自己的数据进行注释。 接下来我们就看看这个包的具体操作。当然了,这类的有监督聚类的R包不止这一个,这里我们只是提供一种思路和演示。 R包链接:https://github.com/powellgenomicslab/scPred/文章链接:scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data | Genome Biology | Full Text本文参考链接:Introduction to scPred • scPred这里我们没有使用示例数据,我们用别的数据进行验证。traning数据集是一个小鼠PMN分好亚群的数据。test数据集是我们自己的小鼠PMN数据。我们利用前面的数据对我们的数据集进行注释。 首先安装R包并对数据进行训练:
library(harmony)
devtools::install_github("powellgenomicslab/scPred")
library(scPred)
library("Seurat")
library("magrittr")
setwd('D:/KS项目/公众号文章/scPred单细胞有监督注释')
#======================================================================================
#===============================1、Reference data training================================
#======================================================================================
reference <- getFeatureSpace(mouse_data, "celltype")
reference <- trainModel(reference)
get_probabilities(reference) %>% head()
# PMN(3) PMN(2) PMN(1) PMN(0)
# Female_NTPH_GCTTTCGGTTCTTCAT-1 4.387384e-01 1.963304e-05 1.408294e-05 0.0001599593
# Female_NTPH_GATGGAGGTCGCTTGG-1 4.119691e-05 8.232246e-01 1.202540e-01 0.0059803552
# Female_NTPH_AGTCTCCAGTCTTCCC-1 1.260117e-04 7.995576e-01 1.583251e-01 0.0002833583
# Female_NTPH_GGAGCAACAGGTCTCG-1 4.984621e-04 7.252164e-01 1.752820e-02 0.0020682616
# Female_NTPH_ATGGATCCAGACACCC-1 5.982061e-04 1.386497e-02 8.812309e-01 0.0023515900
# Female_NTPH_GGGAGTACAGATACCT-1 6.537663e-05 1.787674e-02 9.872667e-01 0.0052899333
# PMN(5) PMN(6) PMN(4) PMN(7)
# Female_NTPH_GCTTTCGGTTCTTCAT-1 0.9226594823 8.498277e-06 5.534320e-07 0.0006438826
# Female_NTPH_GATGGAGGTCGCTTGG-1 0.0002222603 1.723565e-04 6.440098e-06 0.0002707403
# Female_NTPH_AGTCTCCAGTCTTCCC-1 0.0006326606 1.319499e-04 2.916959e-05 0.0003673744
# Female_NTPH_GGAGCAACAGGTCTCG-1 0.1611489503 1.672275e-03 2.831901e-04 0.0006098397
# Female_NTPH_ATGGATCCAGACACCC-1 0.0029780794 1.798155e-04 4.437796e-03 0.0011741416
# Female_NTPH_GGGAGTACAGATACCT-1 0.0074298716 1.011856e-04 1.435643e-05 0.0003051911
get_scpred(reference)
# |Cell type | n| Features|Method | ROC| Sens| Spec|
# |:---------|----:|--------:|:---------|-----:|-----:|-----:|
# |PMN(0) | 1360| 50|svmRadial | 0.998| 0.960| 0.989|
# |PMN(1) | 1295| 50|svmRadial | 0.985| 0.867| 0.970|
# |PMN(2) | 1149| 50|svmRadial | 0.985| 0.862| 0.971|
# |PMN(3) | 745| 50|svmRadial | 0.999| 0.949| 0.995|
# |PMN(4) | 594| 50|svmRadial | 0.996| 0.911| 0.989|
# |PMN(5) | 551| 50|svmRadial | 0.993| 0.869| 0.991|
# |PMN(6) | 267| 50|svmRadial | 0.999| 0.929| 0.998|
# |PMN(7) | 64| 50|svmRadial | 1.000| 0.888| 1.000|
plot_probabilities(reference)
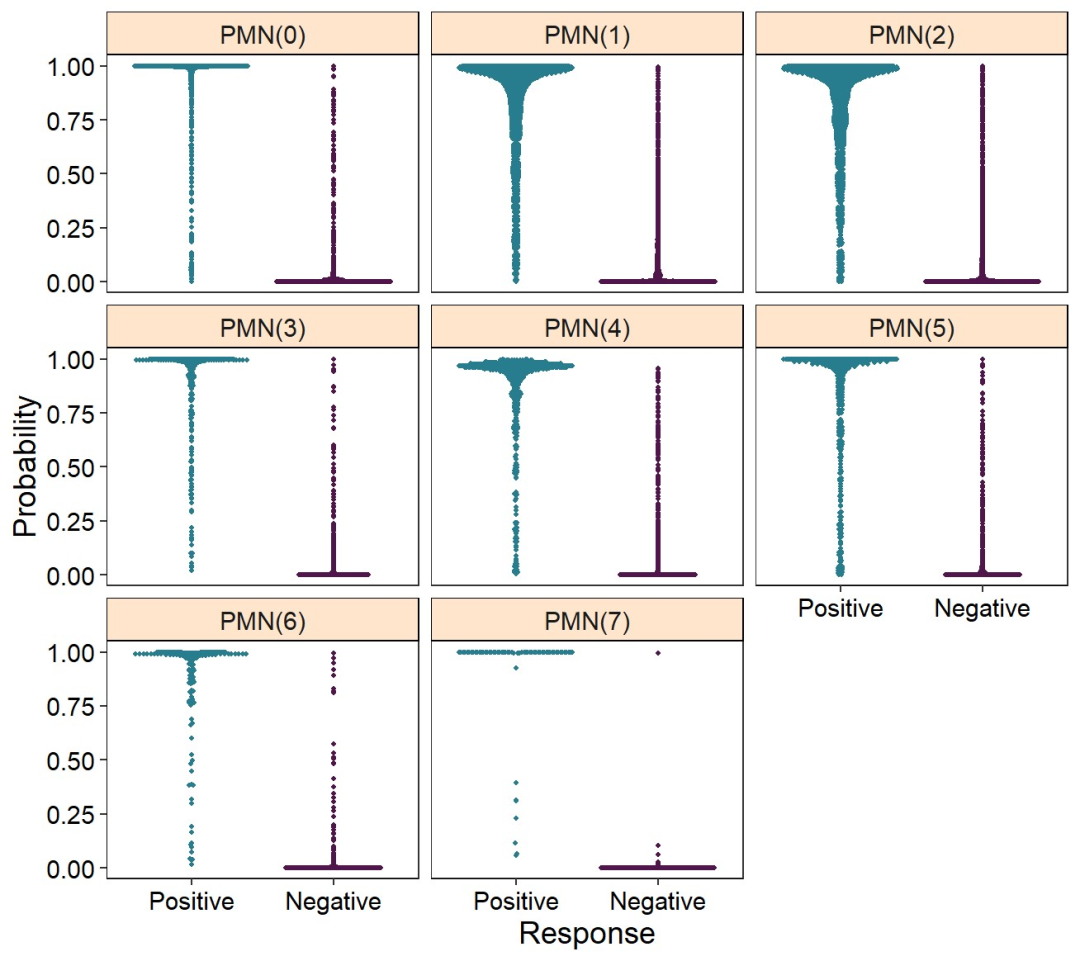
image.png
可以看到训练数据的sens还是可以的。接下来对一个新的数据进行正常的降维分析,然后利用之前的数据集对其注释。
#======================================================================================
#===============================2、Test single cell test================================
#======================================================================================
#做一下标准的降维
BM2 <- read.table('BM2.txt',header = T,row.names = 1)
BM2 <- CreateSeuratObject(counts = BM2, project = "BM2",min.cells = 3, min.features = 200)
BM2[["percent.mt"]] <- PercentageFeatureSet(BM2, pattern = "^mt-")
VlnPlot(BM2, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), pt.size = 0)
#QC一下
BM2 <- subset(BM2, subset = percent.mt < 5)
BM2 <- NormalizeData(BM2)
BM2 <- FindVariableFeatures(BM2, nfeatures = 4000)
Seurat::ElbowPlot(BM2)
BM2 <- ScaleData(BM2, verbose = FALSE)
BM2 <- RunPCA(BM2, npcs = 50, verbose = FALSE)
BM2 <- FindNeighbors(BM2, reduction = "pca", dims = 1:15)
BM2 <- FindClusters(BM2, resolution = 1)
BM2 <- RunUMAP(BM2, reduction = "pca", dims = 1:15)
DimPlot(BM2,label = T)
BM2 <- scPredict(BM2, reference)
DimPlot(BM2, group.by = "scpred_prediction", reduction = "scpred")
BM2 <- RunUMAP(BM2, reduction = "scpred", dims = 1:15)
DimPlot(BM2, group.by = "scpred_prediction", label = TRUE, repel = TRUE)
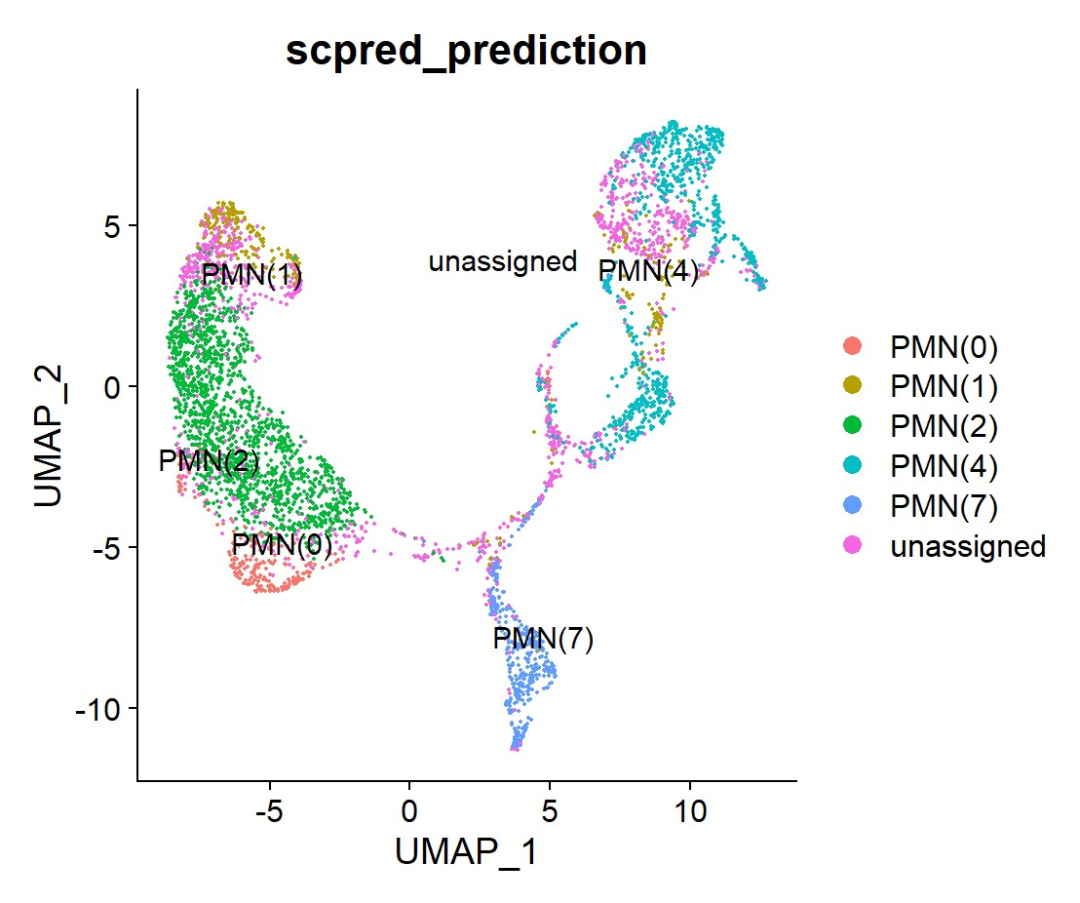
image.png
可以看到有些细胞是没有被识别的,而且PMN3,5,6都没有。我们将未鉴定的细胞去除看看。
PMN <- subset(BM2, scpred_prediction=='unassigned', invert=T)
DimPlot(PMN, group.by = "scpred_prediction", label = TRUE, repel = TRUE)
DimPlot(PMN, label = TRUE, repel = TRUE)
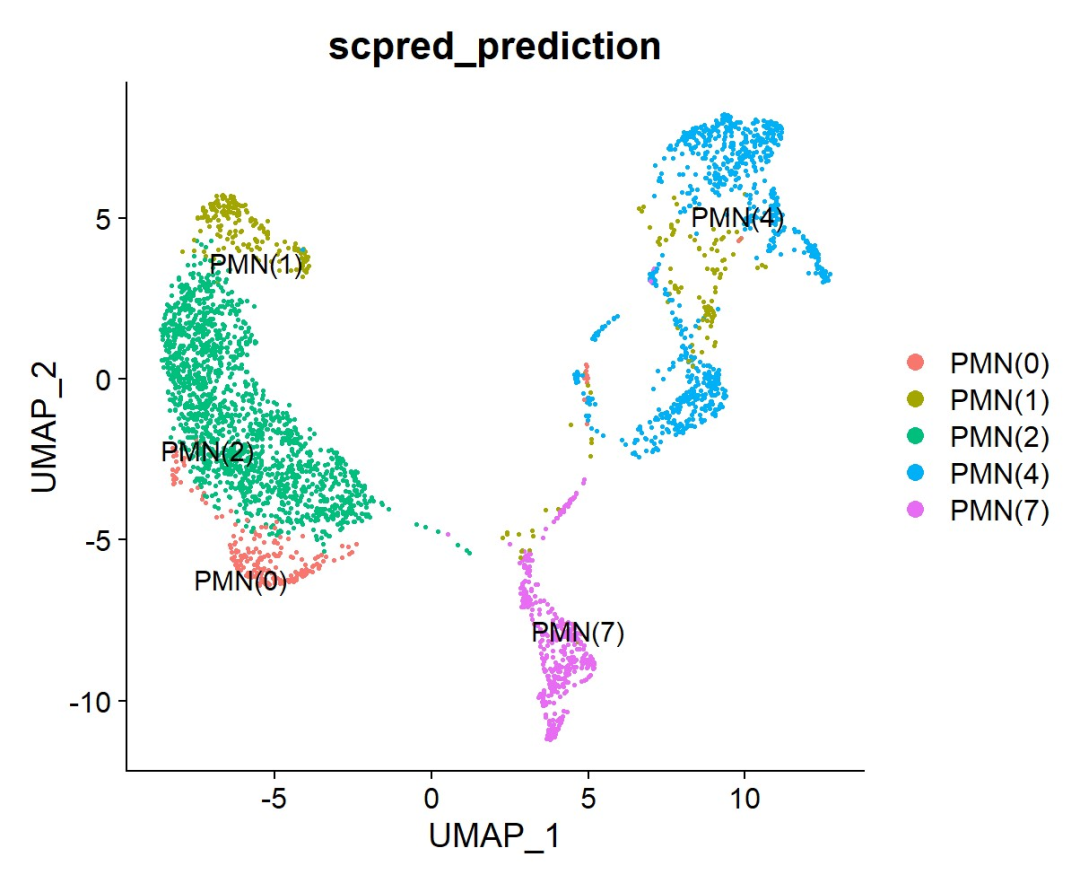
image.png
鉴定到的细胞聚类还是可以的。这里有些细胞没有鉴定到可能的原因是两个数据集的来源不同。不过还是有些细胞群鉴定到了,效果可以的。这里也是提供一种思路,希望对你的研究有帮助。觉得分享有用的点个赞再走呗。

















 943
943

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









