







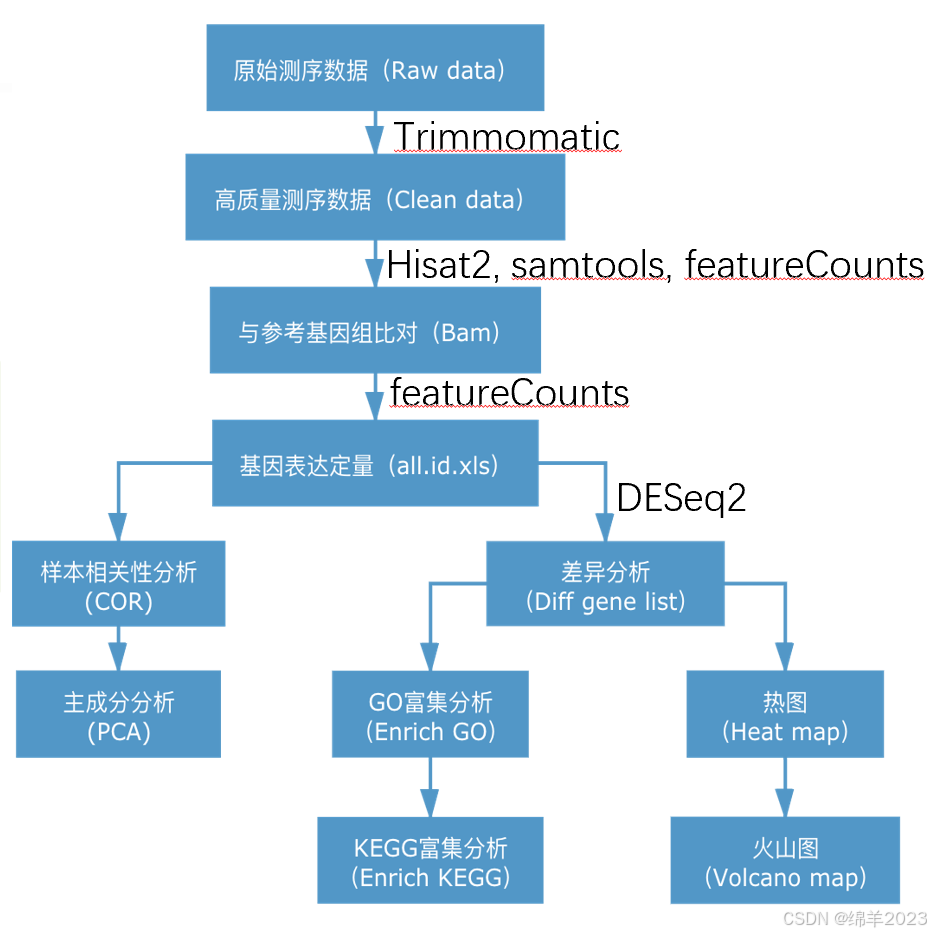
1.准备样本文件
ls *TR_R1.fastq.gz | cut -f 1 -d "_" | sort | uniq > sample_ids.txt #查看左右以TR_R1.fastq.gz结尾的文件,使用cut命令,以“_”为分隔符(-d "_"),选择第一个字段(-f 1),进行排序和去重,写入sample_ids.txt文本文件中。
2. Fastqc先看一下原始数据质量(Fastqc这步可做可不做,可直接做后边的Trimmomatic)
fastqc *fastq.gz -o ./fastqc_initial -t 8
cd ./fastqc_initial/
multiqc .##搜索当前目录,选择FastQC的输出文件,使用8个计算机内核,制作所有样本的汇总html报告。下载网页打开.html文件
3. Trimmomatic修剪
#!/bin/bash
#$ -cwd
#$ -S /bin/bash
#$ -l p=10
cd /data/raw_data
java -jar /data/software/Trimmomatic-0.39/trimmomatic-0.39.jar PE -threads 36 -phred33 B-3HC_R1.fq.gz B-3HC_R2.fq.gz B-3HC_1.clean.fq.gz B-3HC_unpaired1.fq B-3HC_2.clean.fq.gz B-3HC_unpaired2.fq ILLUMINACLIP:/data/software/Trimmomatic-0.39/adapters/TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 #HEADCROP:10 ##是否在质控时就加上“HEADCROP:10”,修剪前10个碱基4. 查看质控结果
mkdir fastqc_trimming #创建一个名为 fastqc_trimming 的新文件夹
fastqc *.fastq.gz -o fastqc_trimming/ -t 10
multiqc .5. 与参看基因组比对
hisat2 -p 10 -x /data/database/genomes/human/hisat2_index/Human_GRCh38_p14_hisat2 -1 ${i}_1.clean.fq.gz -2 ${i}_2.clean.fq.gz -S /data/male/${i}.sam
samtools view -b input.sam | samtools sort -o output.bam #bam文件排序
samtools index -@ 8 -b input.bam #建立bam的索引文件6. 基因定量得到count文件
featureCounts -T 10 -t exon -g gene_id –a /data/database/genomes/human/GCF_000001405.40_GRCh38.p14_genomic.gtf -p -o /data/04.count/${i}_counts.txt ./${i}.bam

















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









